Hematopoietic stem cell transplantation (HSCT) offers potentially curative therapy for patients with thalassemia major and sickle cell disease (SCD). Current myeloablative treatment protocols allow the cure of 78% to 90% of patients with thalassemia and 72% to 96% with SCD, depending on disease status at the time of transplantation. The major limitation to successful transplantation is the lack of a suitable HLA-matched family donor. Unrelated donor HSCT is now extensively used to treat thalassemia, with results similar to those obtained following transplantation using HLA-matched sibling donors. Patients who lack a matched related or unrelated donor can now benefit from successful transplantation using haploidentical donors.
β-Thalassemia and sickle cell anemia (SCA) are among the most widespread single gene disorders worldwide. Globally it is estimated that approximately 7% of the world’s population are carriers of inherited hemoglobin disorders and that 300,000 to 400,000 babies with severe forms of these diseases are born each year. The β-thalassemias are caused by more than 200 different β-globin gene mutations that either reduce or abolish production of the β-globin chain, which lead to the development of ineffective erythropoiesis and hemolytic anemia. The most severe form of disease is characterized by the complete absence of β-globin production and results from the inheritance of 2 β 0 thalassemia alleles, in the homozygous or compound heterozygous states. These combinations usually result in β-thalassemia major; the patients present within 6 months of life with severe anemia and if not treated with regular blood transfusions, die within the first 2 years. The current conventional treatment of β-thalassemia major consists of lifelong regular blood transfusions combined with iron chelation therapy. In developed countries, the optimization of transfusion support and iron chelation therapy have improved the survival of patients with thalassemia converting a previously fatal disease with early death into a chronic, although progressive disease compatible with prolonged survival, eventually for a few decades. Conversely, in less well developed countries where most of these patients reside, most children die before the age of 20 years because of the unavailability of safe blood products and/or expensive iron-chelating drugs. Despite the prolonged life expectancy, a recent study from the United Kingdom Thalassemia Registry showed a steady decline in survival starting from the second decade, with fewer than 50% of patients remaining alive beyond 35 years mainly because of poor compliance with chelation therapy. Allogeneic hematopoietic stem cell transplantation (HSCT) is the only definitively curative therapeutic modality for the treatment of β-thalassemia major. HSCT is a form of gene therapy that uses allogeneic stem cells as vectors for genes essential for normal hematopoiesis. Eventually the vector may well be autologous stem cells transformed by the insertion of normal genes, but there is no indication that this approach will be a widely available clinical option in the foreseeable future. It should be acknowledged that the risk of serious acute and chronic complications of transplantation can present a therapeutic dilemma in light of the observation that, in developed countries where patients with thalassemia receive up-to-date medical treatment, the use of regular blood transfusion and optimal chelation therapy results in a very good survival rate. However, many such patients with increasing age currently experience poor outcomes with conventional treatment, even in countries with universal access to optimal medical treatment.
Patients with SCA have an abnormal hemoglobin (hemoglobin S) that polymerizes under hypoxic conditions, causing increased viscosity, deformation, and membrane damage of the erythrocyte that results in vascular occlusion due to adhesion of sickle cells to each other and to vascular endothelium. The natural history of SCA is characterized by severe chronic hemolytic anemia, acute painful crises, and shortened life-span. Acute stroke, silent cerebral infarcts, acute chest syndrome, and avascular bone necrosis are among the most disabling complications occurring in SCA patients. Several advances such as early penicillin prophylaxis, antipneumococcal vaccine, better blood transfusion practice, and treatment with hydroxyurea in the last 2 decades have contributed both to improved quality of life and to an increase in the life expectancy at least for those patients with access to treatment. Although medical treatments are life-extending, end-organ damage cannot be avoided in most of these patients over time. In fact, a recent prospective longitudinal study over more than 4 decades of observation showed that by the fifth decade of age, nearly 50% of patients had documented irreversible damage of at least one organ. The most common cause of death in children is pneumococcal sepsis, whereas adult patients usually succumb to stroke, multiorgan failure, or acute chest syndrome. At present, HSCT is the only recognized curative therapy for patients with SCA. However, there has been slow progress in the use of HSCT for SCA. Indeed, so far only approximately 250 SCA patients have received HSCT worldwide. The higher individual variability of disease severity, some advances of medical treatment, the lack of universally accepted criteria for transplantation, and the lack of a suitable HLA-identical donor are obstacles for expanding HSCT to many potential candidates. Consequently, HSCT has been mainly offered to pediatric patients with a significant clinical complication such as stroke, recurrent vaso-occlusive events, or anemia requiring chronic blood transfusions. A recent survey among 30 adult patients with sickle cell disease (SCD) about their feelings toward receiving a reduced-intensity bone marrow transplant (BMT) showed that 62% were willing to accept a 10% transplant-related mortality (TRM), one-third of patients were willing to accept a 30% TRM, and 62% accepted a 10% risk of graft failure. Fifty percent accepted infertility, but only 20% considered chronic graft-versus-host disease (GVHD) as an acceptable alternative. Overall, 60% of patients were willing to accept a clinical trial of reduced-intensity BMT. This survey reflects a high morbidity rate among patients with SCD and shows inadequacy of current conservative management strategies to control long-term disease complications. The survey further indicates that many patients with SCD are willing to accept even high toxicities for a potentially curative therapy.
Hematopoietic stem cell transplantation for thalassemia
HSCT Transplantation Tailored to the Patient’s Clinical Condition: Risk Class Approach
In 1988, the authors analyzed the influence of pretransplant characteristics on the outcome of transplantation in 116 consecutive patients aged 16 years or younger who were all treated with exactly the same conditioning regimen and GVHD prophylaxis, starting in June 1985. In a multivariate analysis hepatomegaly and portal fibrosis were associated with a significantly reduced probability of survival, whereas a history of poor compliance with the chelation regimen could not be distinguished from hepatomegaly as a predictor of survival and event-free survival. The influence of pretransplant characteristics on the outcome of transplantation was reexamined in late 1989, by which time 167 patients aged less than 17 years had been treated with the same regimen which, in addition to the previous revealed 2 risk factors, showed a strong statistically significant correlation between a history of iron chelation and survival. The quality of chelation was characterized as regular when desferoxamine therapy was initiated no later than 18 months after the first transfusion and administered subcutaneously for 8 to 10 hours continuously for at least 5 days each week, and any deviation from this regimen was considered as irregular. The degree of hepatomegaly (greater than or not greater than 3 cm), the presence or absence of portal fibrosis in the pretransplant liver biopsy, and the quality of chelation (regular or irregular) given through the years before transplant were identified as variables permitting the categorization of patients into 3 risk classes. Class 1 patients had none of these adverse risk factors, class 3 patients had all 3, and class 2 patients had 1 or 2 adverse risk factors. The results of these studies have allowed the authors to apply treatment protocols tailored to the patient’s clinical condition related to the quality of previous conventional treatment.
The first 2 BMT from HLA-matched sibling donors were performed in Seattle, USA on December 3, 1981 and in Pesaro, Italy on December 17, 1981. The 14-month-old child transplanted in Seattle had received only 250 mL of packed red blood cells (RBCs) at the time of transplantation. The patient had successful engraftment and was cured of thalassemia. The Pesaro approach was based on an assessment that restricting transplants to untransfused patients was impractical, and a 16-year-old thalassemic patient who had received 150 RBC transfusions was given a marrow graft from his HLA-matched brother. This patient rejected the graft and was the first of an extensive series of transplants for thalassemia performed to date.
Experience of the first 20 years (1982–2002)
HSCT in Class 1 and Class 2 Patients
Between 1985 and 1992, 412 class 1 (n = 124) and class 2 (n = 297) patients were treated with busulfan (BU), 3.5 mg/kg/d for 4 consecutive days and cyclophosphamide (CY), 50 mg/kg/d for the subsequent 4 days. As GVHD prophylaxis they received cyclosporine (CSA) and low-dose methylprednisolone. The thalassemia-free survival rates in class 1 and class 2 patients treated during this time frame were 91% and 84%, respectively. The results of transplantation in class 1 and class 2 patients have remained unchanged in subsequent years.
HSCT in Class 3 Patients
Before the risk class approach was adopted, class 3 younger patients (age <17 years) treated with the same preparatory regimen as class 1 and class 2 patients (BU14/CY200) experienced a decreased rate of thalassemia-free survival (53%) and a higher probability of TRM (39%), mainly due to drug-related toxicity. Therefore, the authors reduced the total dose of CY to 120 to 160 mg/kg in the conditioning regimen for class 3 patients to overcome the increased TRM. In fact, this modification improved the probability of overall survival in class 3 younger patients from 53% to 79%, but was associated with an increase in the probability of graft rejection from 7% to 30%, probably due to inadequate immunosuppression and failure to eradicate the massive erythroid hyperplasia characteristic of these patients. The results obtained were not satisfactory and subsequent efforts were made to optimize the treatment protocol for class 3 patients. In April 1997 a new preparatory regimen (Protocol 26) was adopted for class 3 younger patients in an attempt to decrease the higher rejection rate through reducing erythroid expansion and increasing immunosuppression over time to avoid unacceptable peritransplant drug toxicity. This novel treatment regimen involved an intensified preparation with 3 mg/kg of azathioprine and 30 mg/kg hydroxyurea daily from day −45 from the transplant, fludarabine (FLU) 20 mg/m 2 from day −17 through day −13, followed by the administration of BU 14 mg/kg total dose and CY 160 mg/kg total dose. GVHD prophylaxis consisted of CSA, low-dose methylprednisolone, and a modified “short course” of methotrexate (MTX). In addition, patients received hypertransfusion and intravenous chelation and growth factors (granulocyte colony-stimulating factor and erythropoietin) twice weekly to maintain stem cell proliferation in the face of hypertransfusion, thereby facilitating the effect of the hydroxyurea. Importantly this new treatment regimen improved thalassemia-free survival for class 3 patients younger than 17 years from 58% to 85% together with a reduction in the probability of rejection from 30% to 8% as compared with previous preparative regimens.
HSCT in Adult Patients
The authors consider as adult patients those who are 17 years or older. These patients have more advanced disease and treatment-related organ damage, mainly due to prolonged exposure to iron overload and/or hepatitis C. Consequently, most adult patients belong to the class 3 risk group. Since September 1996, 107 adult patients, with a median age of 20 years (range, 17–35 years), received transplants from HLA-identical siblings. There were 18 class 2 patients treated with BU 14 mg/kg and CY 200 mg/kg, and 89 class 3 patients treated with BU 14 to 16 mg/kg and CY 120 to 160 mg/kg as preparatory regimen. An analysis of this cohort of patients showed the probability of survival, thalassemia-free survival, rejection, and nonrejection mortality to be 66%, 62%, 4%, and 37%, respectively. Of note, unlike class 3 younger patients, adult patients treated with the same conditioning regimens had a low graft rejection rate (4%), suggesting that donor-host tolerance might be induced even among extensively transfused patients. This was an unexpected observation indicating that adult patients could benefit from less intensive conditioning regimens with reduced transplant-related toxicity. Therefore, the authors subsequently reduced the total dose of cyclophosphamide to 90 mg/kg in the conditioning regimen for adults and decided to administer to them the preconditioning phase of the Protocol 26 preparatory regimen. A recent analysis of 15 high-risk adult patients treated with this regimen showed a probability of survival, thalassemia-free survival, rejection, and nonrejection mortality of 65%, 65%, 7%, and 28%, respectively.
At present, BMT programs for thalassemia have been established in several countries worldwide, with results similar to those obtained in Pesaro.
Experience of the first 20 years (1982–2002)
HSCT in Class 1 and Class 2 Patients
Between 1985 and 1992, 412 class 1 (n = 124) and class 2 (n = 297) patients were treated with busulfan (BU), 3.5 mg/kg/d for 4 consecutive days and cyclophosphamide (CY), 50 mg/kg/d for the subsequent 4 days. As GVHD prophylaxis they received cyclosporine (CSA) and low-dose methylprednisolone. The thalassemia-free survival rates in class 1 and class 2 patients treated during this time frame were 91% and 84%, respectively. The results of transplantation in class 1 and class 2 patients have remained unchanged in subsequent years.
HSCT in Class 3 Patients
Before the risk class approach was adopted, class 3 younger patients (age <17 years) treated with the same preparatory regimen as class 1 and class 2 patients (BU14/CY200) experienced a decreased rate of thalassemia-free survival (53%) and a higher probability of TRM (39%), mainly due to drug-related toxicity. Therefore, the authors reduced the total dose of CY to 120 to 160 mg/kg in the conditioning regimen for class 3 patients to overcome the increased TRM. In fact, this modification improved the probability of overall survival in class 3 younger patients from 53% to 79%, but was associated with an increase in the probability of graft rejection from 7% to 30%, probably due to inadequate immunosuppression and failure to eradicate the massive erythroid hyperplasia characteristic of these patients. The results obtained were not satisfactory and subsequent efforts were made to optimize the treatment protocol for class 3 patients. In April 1997 a new preparatory regimen (Protocol 26) was adopted for class 3 younger patients in an attempt to decrease the higher rejection rate through reducing erythroid expansion and increasing immunosuppression over time to avoid unacceptable peritransplant drug toxicity. This novel treatment regimen involved an intensified preparation with 3 mg/kg of azathioprine and 30 mg/kg hydroxyurea daily from day −45 from the transplant, fludarabine (FLU) 20 mg/m 2 from day −17 through day −13, followed by the administration of BU 14 mg/kg total dose and CY 160 mg/kg total dose. GVHD prophylaxis consisted of CSA, low-dose methylprednisolone, and a modified “short course” of methotrexate (MTX). In addition, patients received hypertransfusion and intravenous chelation and growth factors (granulocyte colony-stimulating factor and erythropoietin) twice weekly to maintain stem cell proliferation in the face of hypertransfusion, thereby facilitating the effect of the hydroxyurea. Importantly this new treatment regimen improved thalassemia-free survival for class 3 patients younger than 17 years from 58% to 85% together with a reduction in the probability of rejection from 30% to 8% as compared with previous preparative regimens.
HSCT in Adult Patients
The authors consider as adult patients those who are 17 years or older. These patients have more advanced disease and treatment-related organ damage, mainly due to prolonged exposure to iron overload and/or hepatitis C. Consequently, most adult patients belong to the class 3 risk group. Since September 1996, 107 adult patients, with a median age of 20 years (range, 17–35 years), received transplants from HLA-identical siblings. There were 18 class 2 patients treated with BU 14 mg/kg and CY 200 mg/kg, and 89 class 3 patients treated with BU 14 to 16 mg/kg and CY 120 to 160 mg/kg as preparatory regimen. An analysis of this cohort of patients showed the probability of survival, thalassemia-free survival, rejection, and nonrejection mortality to be 66%, 62%, 4%, and 37%, respectively. Of note, unlike class 3 younger patients, adult patients treated with the same conditioning regimens had a low graft rejection rate (4%), suggesting that donor-host tolerance might be induced even among extensively transfused patients. This was an unexpected observation indicating that adult patients could benefit from less intensive conditioning regimens with reduced transplant-related toxicity. Therefore, the authors subsequently reduced the total dose of cyclophosphamide to 90 mg/kg in the conditioning regimen for adults and decided to administer to them the preconditioning phase of the Protocol 26 preparatory regimen. A recent analysis of 15 high-risk adult patients treated with this regimen showed a probability of survival, thalassemia-free survival, rejection, and nonrejection mortality of 65%, 65%, 7%, and 28%, respectively.
At present, BMT programs for thalassemia have been established in several countries worldwide, with results similar to those obtained in Pesaro.
Current experience of HSCT for thalassemia
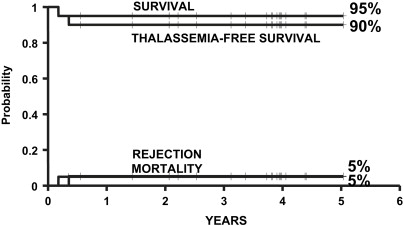
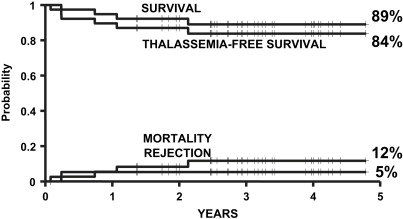
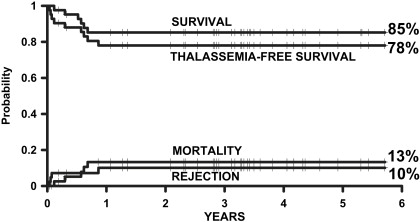
In 2004, the authors of this article and 2 other transplant physicians moved from Pesaro to Rome where they started a new Transplant Program in the International Center for Transplantation in Thalassemia and Sickle Cell Anemia at the Mediterranean Institute of Hematology. Whereas the Pesaro experience was predominantly done on an Italian patient population, the Rome experience was characterized by 2 features: the patients were ethnically very heterogeneous, and the vast majority of these patients was not regularly transfused/chelated, or highly sensitized due to receiving RBC transfusions without the use of leukodepletion filters. Therefore, these patients were likely to have a higher risk of graft rejection as a result of sensitization to HLA antigens. Taking these characteristics into consideration, the authors modified the treatment protocols for transplantation of such patients ( Table 1 ). As of September 2009, 20 class 1, 38 class 2 and 42 class 3 patients, younger than 17 years, received a first HSCT from an HLA-matched family donor at the authors’ institution. Figs. 1–3 show the probability of survival, thalassemia-free survival, rejection, and TRM for class 1, class 2, and class 3 patients, with a median follow-up of 35 months (range, 6–63 months) for surviving patients. The results of this new experience confirmed those obtained in Pesaro, and most importantly, showed the reproducibility of the Pesaro experience in other centers.
| Preconditioning | Conditioning (mg/kg) | GVHD Prophylaxis | |
|---|---|---|---|
| Thalassemia | |||
| Class 1 & class 2 age >4 years | None | ivBu Cy 200 | CSA/MP/short MTX |
| Class 1 & class 2 age ≤4 years | None | ivBu TT 10 Cy 200 | CSA/MP/short MTX |
| Class 3 age <17 years | d −35 to −11 Az, Hu; d −16 to −11 Flu 150 | ivBu TT 10 Cy 160 | CSA/MP/short MTX |
| Adults (age ≥17 years) class 2–3 | d −35 to −12 Az, Hu; d −16 to −12 Flu 150 | ivBu TT 10 Cy 90 | CSA/MP/short MTX |
| Sickle Cell Disease | |||
| None | ivBu Cy 200 ATG 10 | CSA/MP/short MTX | |
HSCT from alternative donors for thalassemia
HSCT from Alternative Related Donors
One of the major obstacles to successful transplantation is the limited number of HLA-matched related donors within families. Approximately 70% of patients lack a suitable family donor. Some of these patients could benefit from HSCT from matched unrelated donors. However, the chance of finding of a matched unrelated donor is strongly dependent on the ethnic background of the patient. The curative potential of HSCT for thalassemia and the limited number of HLA-matched related donors have led to the use of stem cell transplantation from alternative donors in thalassemia. The authors analyzed the results of HSCT in 29 patients with β-thalassemia major who received a marrow graft from HLA phenotypically matched relatives (n = 6), HLA-mismatched siblings (n = 13), parents (n = 8), or relatives (n = 2). Fifteen out of 29 patients received one antigen-mismatched graft. The conditioning regimen consisted of BU/CY in combination with total body irradiation (TBI) or total lymphoid irradiation (TLI). Nine patients also received antilymphocyte globulin. There were high rates of graft failure and TRM (55% and 34%, respectively) and a low thalassemia-free survival rate (21%), showing that these preparative regimens were not adequately myeloablative and immunosuppressive to ensure a high sustained engraftment rate and consequently an adequate probability of disease-free survival.
HSCT from Haploidentical Family Donors
In 2002, the authors’ group started a Haploidentical Transplant program for thalassemia. Patients who lacked an HLA-matched family or unrelated donor were eligible for this study. Between 2002 and 2008, 22 patients with transfusion-dependent thalassemia and median age of 7 years (range, 3–14 years) were given stem cell grafts from haploidentical mothers (n = 20) or brothers (n = 2). All of the patients received an intensified preparative regimen consisting of 3 mg/kg azathioprine and 60 mg/kg hydroxyurea daily from day −59 from the transplant, and fludarabine (FLU) 30 mg/m 2 from day −17 through day −13, aimed at reducing hyperplastic bone marrow and gradually increasing immunosuppression to avoid unacceptable peritransplant drug toxicity. During this time frame, the patients also received hypertransfusion to suppress endogenous erythropoiesis, and granulocyte colony-stimulating factor and erythropoietin to maintain stem cell proliferation, thus facilitating the effect of hydroxyurea. As conditioning regimen, they received BU14/thiotepa (TT)10/CY200 and antithymocyte globulin (ATG). All patients received cyclosporine for GVHD prophylaxis for the first 2 months after transplantation. As stem cell source, 8 patients received T-cell–depleted peripheral blood stem cells (PBSC) (CD34+ immunoselected) and CD3+/CD19+ immunodepleted bone marrow stem cells. The last 14 patients received CD34+ selected peripheral and bone marrow stem cells, using the Clini-MACS procedure. Median infused cell doses were: CD34+ 15.2 × 10 6 /kg, CD3+ 1.8 × 10 5 /kg, and CD19+ 0.27 × 10 6 /kg in the first 8 patients, and CD34+14.2 × 10 6 /kg, CD3+ 2 × 10 5 /kg, and CD19+ 0.19 × 10 6 /kg in the last 14 patients. Overall, 6 patients rejected their grafts, 2 patients died of Epstein-Barr virus–related cerebral lymphoma or cytomegalovirus pneumonia, and 14 patients are alive and cured of thalassemia. The rates of survival, thalassemia-free survival, rejection, and nonrejection mortality were 90%, 61%, 29%, and 14% respectively. In this pediatric patient population, immune reconstitution, especially CD4+ cell recovery, was delayed. These data showed the feasibility of haploidentical stem cell transplantation for patients with thalassemia and, therefore, this procedure should be considered for those younger patients who lack a suitable HLA-matched related or unrelated donor.
Cord Blood Transplantation from Related Donors
Cord blood as an alternative source of hematopoietic stem cells has successfully been used to treat hematological malignant and nonmalignant diseases. The use of umbilical cord blood for transplantation in thalassemia is discussed in greater detail elsewhere in this issue, in the article by Kanathezhath and Walters. A recent study from the Eurocord cooperative group analyzed the outcome of 33 patients with class 1 or class 2 thalassemia, and 11 with SCD who received cord blood cells from a sibling. The majority of patients received BU and CY and ATG, and the remaining patients had BU and FLU as conditioning regimen. In 17 patients TT was added to BU/CY or BU/FLU. Seven (21%) out of 33 patients with thalassemia experienced graft failure. No patient died of transplant-related complications. A low percentage of patients experienced either acute (11%) or chronic (6%) GVHD, which is encouraging. The 2-year estimate of event-free survival was 79%. Although transplanted patients were in the low to intermediate risk classes, the incidence of graft rejection was high. An increased risk of graft failure after cord blood transplantation, compared with that in patients given BMTs, has previously been reported. The one log lower cell dose in cord blood grafts, which is a well known disadvantage, probably has a negative influence on sustained engraftment in recipients with nonmalignant diseases.
HSCT from HLA-Matched Unrelated Donors
At present, many patients who need an allogeneic HSCT and do not have a suitable family donor could benefit from HLA-matched unrelated donor transplantation. Results of HSCT from unrelated donors for the treatment of malignant diseases have improved steadily, mainly due to the introduction of high-resolution molecular techniques for histocompatibility testing and improvements in the management of posttransplant complications. In a recent study, the outcome of BMT from matched unrelated donors prospectively selected using high-resolution molecular typing for HLA class I and class II loci for 68 patients with thalassemia major who received BU/CY or BU/FLU and/or TT as conditioning regimen has been reported. There were 14 class 1, 16 class 2, and 38 class 3 patients. Overall rates of survival, thalassemia-free survival, rejection, and TRM for entire cohort of patients were 79.3%, 65.8%, 14.4%, and 20.7%, respectively. The incidence of acute grade II to IV or grade III to IV GVHD was 40% and 17%, respectively. Ten of 56 evaluable patients (18%) developed chronic GVHD. Class 1 and class 2 patients had much better overall and disease-free survival (96.7% and 80%, respectively) than class 3 patients (65.5% and 54.5%, respectively). These investigators have also recently reported encouraging results in adult patients with thalassemia who received bone marrow from matched unrelated donors, with rates of survival, thalassemia-free survival, TRM, and rejection of 70%, 70%, 30%, and 4%, respectively. Similar results were reported in another study from Asia that included 21 children who received transplants from a matched unrelated donor, as well as 28 patients who received transplants from matched related donors. The 2-year thalassemia-free survival for patients who received transplants from matched unrelated donors was 71%, compared with 82% for patients who received transplants from matched related donors. Although a limited number of patients have been transplanted thus far, these data show that HSCT using HLA-matched unrelated donors, not only in younger but also in adult patients with well-selected donors, may offer success rates similar to those obtained with HLA-identical sibling donors. The main limitation of the experience with matched unrelated donors for HSCT in thalassemia is that, using stringent criteria of HLA matching, only about one-third of thalassemia patients who started the search found a suitable donor in a median time of 3 to 4 months. Using less stringent HLA matching criteria for donor selection, such as allowing 1 or 2 allele mismatches, could increase the donor pool, but the results of such transplants remain to be determined.
Cord Blood Transplantation from Unrelated Donors
Encouraging preliminary results of sibling cord blood transplantation for children with nonmalignant diseases including thalassemia and accumulating experience of successful application of unrelated mismatched cord blood in hematological malignancies and nonmalignant diseases have led to the use of unrelated cord blood transplantation in patients with thalassemia. Recently, the outcome of unrelated cord blood transplantation for 45 patients with nonmalignant diseases, with a median age of 4.5 years (range, 0.1–16.2 years), has been reported. Cord blood units were matched at 4 (or more) out of 6 HLA-A, HLA-B, and HLA-DRB1 loci. There were 32 transfusion-dependent thalassemia patients, 21 with class 1 characteristics and 9 with class 2 characteristics (2 patients were unclassified). The conditioning regimen for these patients consisted of intravenous busulfan, 14 mg/kg total dose and cyclophosphamide, 200 mg/kg total dose. Five patients (16%) had primary (n = 3) or secondary (n = 2) graft failure and all 5 had sustained engraftment following a second transplantation. The incidence of grade II to IV and grade III to IV acute GVHD was 76% and 42%, respectively. The overall incidence of chronic GVHD was 35%, with only 1 patient experiencing extensive disease. TRM at 2 years was 12%. The results of this study are encouraging, and the high rates of graft rejection and of grade III to IV acute GVHD observed in these patients could probably be reduced by using better matched (5/6 or 6/6) unrelated cord blood units.
Mixed Chimerism Following HSCT for Thalassemia
Mixed hematopoietic chimerism (MC) is an interesting phenomenon after HSCT for thalassemia. The authors’ group has evaluated MC in 335 patients who received BMT from HLA-matched family donors for thalassemia. The incidence of MC at 2 months after transplant was 32.2%. Of the 227 patients with complete donor chimerism, none rejected their grafts while 35 of 108 patients (32.4%) with MC lost the graft, indicating that MC is a risk factor for graft rejection in patients with thalassemia. The percentage of residual host hematopoietic cells (RHCs) determined at 2 months after transplant was predictive for graft rejection, with nearly all patients experiencing graft rejection when RHCs exceeded 25%, and only 13% of patients having graft rejection when RHCs were less than 10%. The risk of rejection for patients who had RHCs between 10% and 25% was 41%.
Unlike hematological malignancies in which residual host cells are predictive of relapse, patients with thalassemia can have lifelong stable mixed chimerism without rejection following transplantation. Ten percent of patients receiving BMT for thalassemia following myeloablative conditioning became persistent mixed chimeras and became transfusion-independent, suggesting that a limited number of engrafted donor cells might be sufficient to provide significant improvement of disease phenotype in patients with thalassemia major once donor-host tolerance has been established.
Related posts:
 HbE/β-Thalassemia: Basis of Marked Clinical Diversity
HbE/β-Thalassemia: Basis of Marked Clinical Diversity
 Iron Overload in Thal assemia and Related Conditions: Therapeutic Goals and Assessment of Response to Chelation Therapies
Iron Overload in Thal assemia and Related Conditions: Therapeutic Goals and Assessment of Response to Chelation Therapies
 Lower Gastrointestinal Tract Cancer Predisposition Syndromes
Lower Gastrointestinal Tract Cancer Predisposition Syndromes
 Anemia, Ineffective Erythropoiesis, and Hepcidin: Interacting Factors in Abnormal Iron Metabolism Leading to Iron Overload in β-Thalassemia
Hemoglobin Gene Therapy for β-Thalassemia
Mouse Models of Inherited Cancer Syndromes
Anemia, Ineffective Erythropoiesis, and Hepcidin: Interacting Factors in Abnormal Iron Metabolism Leading to Iron Overload in β-Thalassemia
Hemoglobin Gene Therapy for β-Thalassemia
Mouse Models of Inherited Cancer Syndromes
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


