Fig. 7.1
Five-year cumulative incidence of HCC in different etiologies of liver cirrhosis (Data from Fattovich et al., Gastroenterology 2004;127:S35-50)
Obviously, structural and functional abnormalities pertinent to alcoholic cirrhotic transformation such as profound alterations of extracellular matrix composition, growth factor and cytokine milieu, disturbed angiogenesis, impaired immune function and reduced capacity of cirrhotic tissue to handle oxidative and/or toxic insults result in an environment that favours dedifferentiation and malignant growth.
Certain histological features typically seen in established HCC are already present, albeit to a lesser extent, in alcoholic cirrhosis and thereby support the assumption that some pathogenic hallmarks leading to cirrhosis precede those causing HCC [6]. Enzyme-altered foci and preneoplastic nodules are typically observed premalignant lesions which can also be induced in experimental rodent models of HCC [18]. Interestingly, Mallory body (MB) formation is high in HCC, and the incidence of HCC is significantly higher in cirrhosis with MBs than without leading to the hypothesis that MBs reflect an initial phenotypical alteration in the carcinogenic transformation of hepatocytes [19]. In addition, oval cells—pluripotent liver progenitor cells—are present in premalignant liver tissues HCC and adjacent tissues and evolve in response to long-term alcohol exposure [20].
HCC evolution is closely linked to chronic liver injury from various causes requiring cell/tissue regeneration for replacement of hepatocyte decay, but rarely develops in healthy liver during physiological aging. One possible explanation for this tight correlation is that HCC development requires increased cell proliferation, leading to the stepwise accumulation of genetic hits allowing for dysplastic and eventually malignant transformation. The most common and unifying condition associated with hepatocarcinogenesis is cirrhosis which takes long to develop (20–40 years). As mentioned above, cirrhosis induces alterations of the microenvironment including altered cytokine secretion from activated hepatic stellate cells and portal fibroblasts, as well as proinflammatory stimuli from invading immune cells. Regarding the latter, molecular signals derived from proinflammatory tumour-necrosis factor α (TNFα) are considered pivotal in ALD [13]. Excessive alcohol consumption can lead to an increased portosystemic uptake of lipopolysaccharides/endotoxins from gram-negative gut bacteria which contribute to necroinflammation and fibrosis progression via various molecular mechanisms including TNFα and the CD14/toll-like receptor-4 complex to produce ROS via NADPH oxidase [21–23]. In fact, elevated TNFα levels and other proinflammatory cytokines are prominent features of ALD, resulting in hepatocyte proliferation or apoptotic/necrotic death, recruitment of inflammatory cells and tissue remodelling. Molecular responses are triggered upon binding of TNFα to its cellular receptors on hepatocytes and other liver cells leading to activation of adaptor protein 1 (AP-1; c-jun/c-fos), crosstalk with epidermal growth factor signalling and subsequently enhanced cell proliferation and potentially to apoptosis via caspase activation [24]. Beyond that, TNFα activates sphingomyelinase to increase intracellular ceramide which inhibits the mitochondrial electron transport chain. Consequently increased production of ROS promotes lipid peroxidation and apoptosis independently of caspases. However, increased oxidative stress also contributes to the activation of transcription nuclear factor κB which is instrumental for the initiation of cell survival mechanisms involving the upregulation of antiapoptotic proteins such as Bcl-2, manganese superoxide dismutase and nitric oxide synthase that can all protect mitochondrial integrity and function. Indeed, constitutive upregulation of nuclear factor κB expression has been convincingly demonstrated both in human and experimental ALD [25, 26] and is a typical feature in human HCC [27]. Hence, TNFα may dose-dependently activate cellular survival mechanisms or precipitate apoptosis and/or necrosis. This may provide an explanation why hepatocytes challenged by inflammatory insults below the threshold of cell death may acquire proliferative advantages and become susceptible to dedifferentiation triggered by carcinogens such as (alcohol-derived) acetaldehyde.
7.4 Alcohol as a Risk Factor for HCC in Non-alcoholic Liver Diseases
Chronic alcohol consumption is an established risk modifier of HCC development in patients with other, non-alcoholic concomitant liver diseases, particularly viral hepatitis [28], hereditary hemochromatosis (HH) [29] and non-alcoholic fatty liver disease (NAFLD) [30]. Hepatitis B and C infections account for the large majority of HCC cases in the developing world, whereas NAFLD along with the obesity epidemic is an increasing cause of HCC in Western countries. In these diseases, which render the liver susceptible to additional oncogenic insults, chronic alcohol consumption even at moderate levels could have a striking influence on the population-based risk of HCC risk [31].
7.4.1 Coexisting Chronic Viral Hepatitis
A large Italian case-control study including 464 subjects with HCC as cases and 824 subjects without liver diseases as controls demonstrated a synergism between alcohol drinking (>60 g/day) and hepatitis B or C virus infection regarding the risk of HCC with approximately a twofold increase in the odds ratio for each viral infection for drinkers [32] (Fig. 7.2).
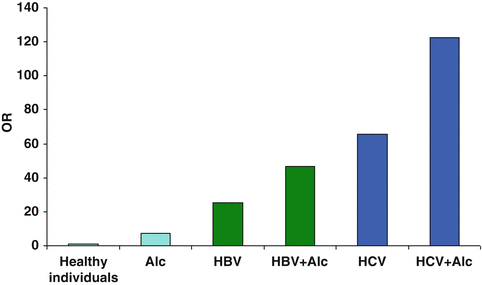
Fig. 7.2
Odds ratios (OR) for HCC in drinkers with/without chronic viral hepatitis B or C. Coexisting alcohol drinking doubles the risk of HCC in patients infected with hepatitis B or C virus (Data from Donato et al. Am J Epidemiol 2002;155:323–31)
Not surprisingly, the coexistence of two liver diseases (alcohol + chronic infection with hepatitis viruses) synergistically enhances the risk of liver disease progression and also HCC. However, the mechanisms leading to HCC evolution are imprecisely defined and may be distinct between the two types of viral hepatitis.
7.4.2 Hepatitis B
Data from human studies on the interaction between HBV infection and alcohol consumption are scarce. Marcellin and co-workers estimated the mortality related to HCV and HBV infections in France in 2001. They found that 95 % of patients with HCV infection who died had cirrhosis, and 33 % had HCC, similar to the figures in the HBV infection group in which 93 % of subjects had cirrhosis and 35 % had HCC. Overall, deaths related to either HBV or HCV infection occurred at an earlier age in patients with a history of excessive alcohol consumption [30]. In a similar study from Italy based on data from the Dionysos study, authors quantified the burden of chronic viral liver disease and the role of alcohol intake to morbidity and mortality in a population-based study. Here, ethanol intake was found to be an independent predictor of cirrhosis in subjects with chronic HCV infection and an independent predictor of death in subjects with either HCV or HBV infection [33]. Comparable data come from a prospective cohort study in Taiwan which demonstrated that habitual alcohol consumption can increase the risk of HCC in HBsAg-positive individuals with an odds ratio of 1.86 (1.32–2.61) [34]. A population-based cohort study from Korea found that in the subgroup of chronic HBV carriers, the HCC risk rose dose dependently with an alcohol intake of 50–99 g/day with a relative risk of 1.2 (95 % CI 1.0–1.5) and of 1.5 (95 % CI 1.2–2.0) for >100 g/day [35]. Whether this synergistic effect on the risk of HCC from alcohol and coexistent HBV infection is additive or exponential is not known.
Putative mechanisms are yet unknown, but may relate to a distinct pattern of epigenetic regulation of certain HCC-associated genes as evidenced by Lambert et al. who showed a high frequency of aberrant hypermethylation of specific genes (RASSF1A, GSTP1, CHRNA3 and DOK1) in HCCs compared to control cirrhotic or normal liver tissues [36]. An association between alcohol intake and hypomethylation of the methyl-guanine methyltransferase gene promoter was demonstrated, whereas HBV infection was linked to promoter hypermethylation of glutathione S-transferase, indicating that hypermethylation of the genes analysed in HCC tumours exhibits remarkably distinct patterns between associated risk factors.
7.4.3 Hepatitis C
Ample evidence exists demonstrating a clear synergistic effect between coexisting alcohol abuse and chronic HCV infection with regard to liver disease progression and HCC development. This is particularly important since the prevalence of HCV infection is significantly higher among heavy drinkers than in the general population; for example, while HCV antibody positivity in the general population of the USA is approximately 1 %, this figure increases to 16 % among alcoholics and reaches even 30 % in alcoholic individuals with liver disease [37–39].
A large population-based study from Northern Italy analysed risk factors of progression of chronic hepatitis C and development of HCC in anti-HCV-positive subjects derived from the Dionysos cohort and found a daily alcohol consumption of >90 g/day to be a significant risk factor for HCC [40]. Hassan and co-workers conducted a hospital-based, case-control study among 115 HCC patients and 230 non-liver cancer controls matched for age, sex and year of diagnosis [41]. Factors independently associated with HCC were chronic hepatitis B and C, alcohol consumption (>80 g/day) and type II diabetes. Significant synergism was observed between heavy alcohol consumption and chronic HCV infection (OR 53.9; 95 % CI 7.0–415.7) and diabetes mellitus (OR 9.9; 95 % CI, 2.5–39.3). The study underlines that heavy alcohol consumption contributes to the magnitude of HCC cases (32 %), whereas 22 %, 16 % and 20 % were explained by HCV, HBV and diabetes mellitus, respectively. Similar data have been gathered for Europe and Asia as well in which concomitant alcohol consumption in HCV-infected individuals increases the risk of HCC additively, if not exponentially [42, 43].
The underlying pathophysiology of this synergistic impact on HCC evolution is still not completely understood but may relate to joint effects of both alcohol and HCV on certain effects conveyed by HCV epitopes on key molecular events instrumental in hepatocarcinogenesis. In keeping, experimental evidence generated by Moriya and associates is highly suggestive of a direct oncogenic effect of the HCV core protein in mice [44]. In their study, the development of HCC in two independent lines of transgenic mice overexpressing the HCV core gene was reported. The same mice spontaneously developed steatosis early in life as a feature of chronic hepatitis C infection and of alcohol, but not more advanced liver disease (Fig. 7.3). The latter similarity allows for speculations with regard to coexisting alcohol abuse [45]: the downstream events triggered by the core protein are segregated into two components. One is the augmented production of oxidative stress along with the activation of scavenging system, including catalase and glutathione, in the putative preneoplastic stage with steatosis in the liver. Thus, oxidative stress production in the absence of inflammation by the core protein would partly contribute to the development of HCC. The generation of oxidative stress is estimated to originate from mitochondrial dysfunction in hepatocytes by HCV infection. Obviously, oxidative stress from concomitant alcohol consumption would further intensify these HCV-related effects. The other component is the alteration of intracellular signalling cascade of mitogen-activated protein kinase (MAPK) and activating factor (AP)-1, leading to the activation of cell growth and proliferation. Notably, AP-1 upregulation is a key observation in alcohol-mediated liver cell regeneration via retinoic acid receptors, and MAP kinase cascades and their regulation by the phosphoinositide-3-kinase/Akt signalling cascade appear to be crucial in the onset of alcohol-mediated cell injury [46]. These mechanisms are also hallmarks of alcohol-associated hepatocarcinogenesis, and joint impact from both alcohol and HCV on these molecular targets represents a potential explanation for the increased incidence of HCC in HCV-infected alcoholics.
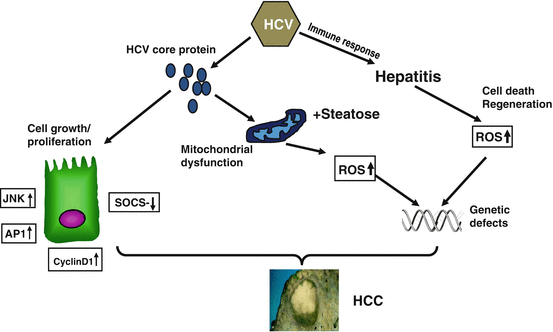
Fig. 7.3
HCV core protein promotes hepatocarcinogenesis indirectly via causing chronic inflammation and mitochondrial dysfunction. However, evidence exists that HCV may also directly cause malignant transformation through stimulating hyperproliferation, reduction of cytokine release and upregulation of cell survival mechanisms
7.5 Non-alcoholic Fatty Liver Disease
Since hepatic histological changes in NASH as well as in alcoholic steatohepatitis (ASH) are strikingly similar, and regular alcohol consumption in obese subjects is rather the rule than the exception, it is not surprising that alcohol consumption may aggravate obesity-related liver disease and vice versa.
Accordingly, two studies from France demonstrated that alcohol consumption is a confirmed risk factor of steatosis and fibrosis progression in obese individuals [47, 48]. A more recent study from Sweden investigated whether low alcohol intake, consistent with the diagnosis of NAFLD, is associated with fibrosis progression in 71 subjects with biopsy-proven NAFLD [49]. By multivariate binary logistic regression analysis, heavy episodic drinking (p < 0.001) and insulin resistance (p < 0.01) were independently associated with significant fibrosis progression.
These results contrast with a recent cross-sectional analysis of adult participants in the NIH NASH Clinical Research Network, which studied only modest or non-drinkers, while drinkers with heavy or otherwise harmful drinking behaviour were excluded. This study surprisingly showed that modest drinkers (daily alcohol consumption <20 g) compared to non-drinkers had lower odds of having NASH (summary odds ratio 0.56, 95 % CI 0.39–0.84, p = 0.002) and also lesser fibrosis (OR 0.56 95 % CI 0.41–0.77) and hepatocellular ballooning (OR 0.66 95 % CI 0.48–0.92) than lifetime non-drinkers [50]. Cross-sectional data from Japan support this finding by demonstrating a significant inverse correlation between drinking frequency and the prevalence of fatty liver (p < 0.001) both in men and women [51]. Thus, a total embargo on any alcohol consumption in patients with NASH is currently not supported by human data.
However, very few data exist focussing on the interaction between features of the metabolic syndrome associated with NAFLD and concomitant alcohol consumption regarding HCC development. However, bearing in mind that both NAFLD and ALD are independent risk factors of HCC, coincidence of either etiology in an individual will likely increase the risk of HCC, particularly when considering that the mechanisms leading to HCC in either disease are remarkably similar (Fig. 7.4).
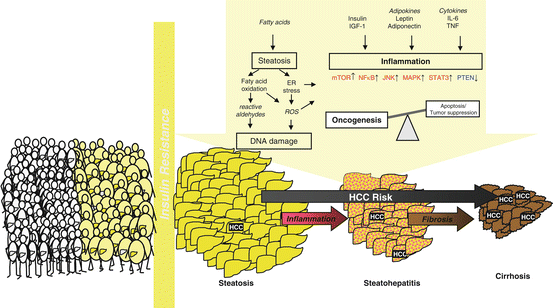
Fig. 7.4
Pathophysiology of alcoholic and non-alcoholic fatty liver disease shares many similarities which synergistically promote HCC development in subjects who harbour both risk factors as a result of their lifestyle (Adapted from Stickel F, Hellerbrand C. Gut 2010;59:1303–7)
7.6 Alcohol-Mediated Activation of Environmental Carcinogens
Alcoholics may be exposed to carcinogens or procarcinogens ingested along with alcoholic beverages which may contain nitrosamines, polycyclic hydrocarbons, asbestos fibres and fusel oils [52]. In addition, many alcoholics are smokers and epidemiological surveys have shown a hyperadditive effect of alcohol and smoking in increasing the risk of developing HCC [53]. Some of these procarcinogens are activated by cytochrome P450 2E1 (CYP2E1), which is induced by chronic ethanol consumption (see below). Thus, potent hepatocarcinogens, such as nitrosamines, aflatoxins as well as vinyl chloride, are substrates of CYP2E1 and undergo activation [54].
Importantly aflatoxin B1 can induce a mutation in codon 249 of the p53 tumour suppressor gene which is frequently found in human HCC [55]. Although animal experiments have been controversial as to whether ethanol enhances AFB1-induced hepatocarcinogenesis, an epidemiological study on AFB1 exposure demonstrated that even a moderate daily consumption of 24 g ethanol increases the risk of developing HCC induced by 4 μg of dietary AFB1 by 35-fold [56].
7.7 Ethanol Metabolism and HCC
More than 90 % of ethanol is metabolized in the liver, either by alcohol dehydrogenase (ADH), the microsomal ethanol oxidizing system (MEOS) represented by CYP2E1 or to a much smaller extent by catalase. While catalase plays only a minor role in hepatic ethanol degradation, ADH and MEOS produce substantial amounts of acetaldehyde (AA), the first and most toxic metabolite of ethanol as well as reactive oxygen species (ROS), of which both may contribute to hepatocarcinogenesis.
7.8 Acetaldehyde
Acetaldehyde (AA) is a recognized cancer-inducing agent due to its toxicity, mutagenicity and carcinogenicity according to a consensus of the International Agency for the Research on Cancer (IARC) [9]. AA interferes with DNA synthesis and DNA repair. In vivo and in vitro experiments in prokaryotic and eukaryotic cell cultures as well as in animal models have provided evidence that AA has direct mutagenic and carcinogenic effects. AA causes point mutations in the hypoxanthine-guanine-phosphoribosyltransferase locus in human lymphocytes and induces sister chromatid exchanges and gross chromosomal aberrations [57–61]. AA directly inhibits O6-methylguanosyl transferase, an enzyme that repairs DNA adducts [62].
Most importantly, however, AA binds to DNA and forms stable adducts [61, 63–69]. Binding to DNA represents one mechanism by which AA could trigger replication errors and/or mutations in oncogenes and tumour suppressor genes. It has been shown that the major stable DNA adduct N2-ethyl-desoxyguanosine (N2-Et-dG) serves as a substrate of eukaryotic DNA polymerase. However, N2-Et-dG seems rather a marker for chronic ethanol consumption than a major risk lesion for cancer. In addition, another DNA adducts of AA, 1,N2-propano-desoxyguanosine (PdG) has been identified, especially in the presence of basic amino acids, histones and polyamines. While N2-Et-dG is non-mutagenic and may represent a marker of chronic alcohol ingestion, PdG has mutagenic properties.
Evidence of the causal role of AA in ethanol-mediated carcinogenesis comes from genetic candidate gene case-control studies in alcoholics. Individuals who accumulate AA due to polymorphisms and/or mutations in the genes coding for enzymes responsible for AA generation and degradation have been shown to have an increased cancer risk. Thus, individuals who drink alcohol and have a deficient aldehyde dehydrogenase (ALDH) such as 40 of the Asian population with increased AA levels after drinking also have a high risk for various cancers such as those of the upper aerodigestive tract and the colon [70]. Similarly individuals who produce more AA due to a rapid alcohol dehydrogenase (ADH1C1*1) also have an increased risk for these cancers including HCC [71].
7.9 Oxidative Stress
Oxidative stress by the formation of ROS such as superoxide anion and hydrogen peroxide is another hallmark of alcohol-mediated tissue injury. Several enzyme systems are capable to produce ROS, including CYP2E1, the mitochondrial respiratory chain and the cytosolic enzymes xanthine oxidase and aldehyde oxidase [72]. Ethanol-mediated ROS formation may be due to an increased electron leakage from the mitochondrial reparatory chain associated with the stimulation of reduced nicotinamide adenine dinucleotide (NADH) shuttling into mitochondria and to the interaction between N-acetylsphingosine (via TNF-α) and mitochondria. The induction of sphingomyelinase by TNF-α increases the levels of ceramide, an inhibitor of the activity of the mitochondrial electron transport chain, leading to increased mitochondrial production of ROS [73]. ROS can also be generated in alcoholic hepatitis with activated hepatic phagocytes [74]. Hepatic iron overload as observed in alcoholic liver disease increases ROS and finally nitric oxide production due to ethanol-mediated stimulation of inducible nitric oxide synthase results in the formation of highly reactive peroxynitrite [75].
Most important in ALD, however, is the production of ROS via alcohol-induced CYP2E1. This induction is an adaptive process and associated with an increased metabolism of ethanol to acetaldehyde and ROS. Most likely, increased CYP2E1 activity results from an inadequate degradation of CYP2E1 by the ubiquitin proteasome pathway impaired by alcohol. A significant increase in hepatic CYP2E1 activity may already occur following the ingestion of 40 g of ethanol daily for 1 week [76]. In animal experiments, the induction of CYP2E1 correlates with NAD phosphate (NADPH) oxidase activity, the generation of hydroxyethyl radicals, lipid peroxidation and the severity of hepatic damage, all of which could be prevented by the CYP2E1 inhibitor chlormethiazole [77]. In addition, premalignant DNA lesions were found more frequently in CYP2E1 wild-type mice compared to animals knocked out for CYP2E1 [78], and transgenic mice overexpressing CYP2E1 had more severe liver injury [79].
ROS produced by CYP2E1 results in lipid peroxidation. Various lipid peroxidation products including 4-hydroxynonenal may bind to purine and pyrimidine bases thereby forming exocyclic DNA adducts which can be mutagenic and carcinogenic [80, 81]. Biopsies from patients with various degrees and severities of alcoholic liver disease reveal a gradual increase of exocyclic DNA adducts along with the degree of alcoholic liver injury and a significant correlation with 4-HNE immunostaining and CYP2E1 induction [82]. This takes already place at the stage of alcoholic fatty liver [83]. Notably, the formation of etheno-DNA adducts can be inhibited by treatment with the CYP2E1 inhibitor chlormethiazole.
7.10 Alcohol and Altered DNA Methylation
Apart from genetic changes along with chronic alcoholism, i.e. mutations, DNA cross links or impaired DNA repair, chronic and acute alcohol intake may also affect epigenetic mechanisms of gene expression such as methylation of DNA. DNA methylation is an important determinant in controlling gene expression whereby hypermethylation has a silencing effect on genes and hypomethylation may lead to increased gene expression. And indeed, alcohol intercepts with these epigenetic mechanisms [84].
Alcohol interacts with absorption, storage, biologic transformation and excretion of compounds which are essential for methyl group transfer including folate, vitamin B6 and certain lipotropes. Especially, the production of S-adenosyl-l-methionine (SAMe), the universal methyl group donor in methylation reactions, is impaired. Furthermore, ethanol compromises SAMe synthesis through inhibition of crucial enzymes involved in SAMe generation. This can impair the formation of endogenous antioxidants such as glutathione and also lead to reduced cellular membrane stability [85].
In addition, alcohol interacts with the methylation status of certain genes and thereby contributes to liver damage and tumour development. Accordingly, alcohol-induced depletion of lipotropes may cause hypomethylation of oncogenes leading to their activation. The decrease in methylation capacity caused by chronic alcohol consumption can therefore contribute to epigenetic alterations of genes involved in hepatocarcinogenesis.
7.11 Interaction of Alcohol with Retinoids
It has been shown for decades that chronic alcohol consumption lowers hepatic vitamin A levels, especially in advanced alcoholic liver disease [86]. Retinoic acid (RA) plays an important role in controlling cell growth differentiation, apoptosis and is of potential clinical interest in cancer prevention and treatment. Therefore, interaction of ethanol with RA metabolism has important implications on the evolution, prevention and treatment of alcohol-related cancers.
The mechanism of alcohol-associated decrease in retinol and RA has multiple causes (Fig. 7.5). Since ADH and ALDH share the common substrates ethanol and retinol as well as AA and retinal to form RA, an interaction at these enzyme sites is not surprising. It has been demonstrated that ethanol acts as a competitive inhibitor of retinol oxidation [87]. Beside the fact that ethanol competes with retinol for the binding site of ADH, there are other mechanisms explaining the decrease in RA. Since chronic ethanol consumption increases CYP2E1 activity, an enhanced catabolism of vitamin A and RA into polar metabolites due to an induction of cytochrome P-450 2E1 occurs [87]. Although a variety of cytochrome isoenzymes such CYP1A1, CYP2B4, CYP2C3, CYP2C7, CYP2E1 and CYP26 are involved in RA metabolism, CYP2E1 seems of major importance [87]. The involvement of CYP2E1 in the metabolism of RA was proven by the fact that the CYP2E1 inhibitor chlormethiazole can completely inhibit this degradation [88]. The prevention of reduced RA status in the liver of ethanol-fed rats by chlormethiazole treatment indicates that the breakdown of retinoids by microsomal CYP2E1 is a key mechanism for the ethanol-enhanced catabolism of retinoids in hepatic tissue after treatment with alcohol. Chronic ethanol consumption with low hepatic RA concentrations results in a downregulation of RA receptors and an up to eightfold increased expression of the AP-1 (c-jun and c-fos) transcriptional complex [87]. This explains hepatocellular hyperproliferation as AP-1 is a central complex downstream of various growth factors, oncogenes and tumour promoters. Supplementation of RA to animals not only results in a decrease of AP-1 gene expression but also in reduced hepatic proliferation.
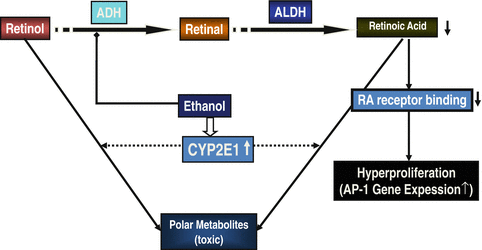
Fig. 7.5
 A Perspective on Chemoprevention by Resveratrol in Head and Neck Squamous Cell Carcinoma
Transgenic Mouse Models for Alcohol Metabolism, Toxicity, and Cancer
A Perspective on Chemoprevention by Resveratrol in Head and Neck Squamous Cell Carcinoma
Transgenic Mouse Models for Alcohol Metabolism, Toxicity, and Cancer
 Genetic Polymorphisms of Alcohol Dehydrogense-1B and Aldehyde Dehydrogenase-2, Alcohol Flushing, Mean Corpuscular Volume, and Aerodigestive Tract Neoplasia in Japanese Drinkers
Genetic Polymorphisms of Alcohol Dehydrogense-1B and Aldehyde Dehydrogenase-2, Alcohol Flushing, Mean Corpuscular Volume, and Aerodigestive Tract Neoplasia in Japanese Drinkers
 Alcohol Consumption, Wnt/β-Catenin Signaling, and Hepatocarcinogenesis
Alcohol Consumption, Wnt/β-Catenin Signaling, and Hepatocarcinogenesis
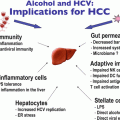 Alcohol and HCV: Implications for Liver Cancer
Alcohol and HCV: Implications for Liver Cancer
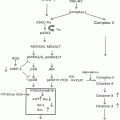 Synergistic Toxic Interactions Between CYP2E1, LPS/TNFα, and JNK/p38 MAP Kinase and Their Implications in Alcohol-Induced Liver Injury
Synergistic Toxic Interactions Between CYP2E1, LPS/TNFα, and JNK/p38 MAP Kinase and Their Implications in Alcohol-Induced Liver Injury




Interaction between alcohol and retinoid turnover takes place at various steps and results in a depletion of retinoic acid (RA) which augment hyperproliferation. In addition, alcohol-induced CYP2E1 causes the formation of polar metabolites from RA contributing to hepatocellular damage
Related posts:
A Perspective on Chemoprevention by Resveratrol in Head and Neck Squamous Cell Carcinoma
Transgenic Mouse Models for Alcohol Metabolism, Toxicity, and Cancer
Genetic Polymorphisms of Alcohol Dehydrogense-1B and Aldehyde Dehydrogenase-2, Alcohol Flushing, Mean Corpuscular Volume, and Aerodigestive Tract Neoplasia in Japanese Drinkers
Alcohol Consumption, Wnt/β-Catenin Signaling, and Hepatocarcinogenesis
Alcohol and HCV: Implications for Liver Cancer
Synergistic Toxic Interactions Between CYP2E1, LPS/TNFα, and JNK/p38 MAP Kinase and Their Implications in Alcohol-Induced Liver Injury
Stay updated, free articles. Join our Telegram channel
Full access? Get Clinical Tree
