Fig. 17.1
CEA processing and metastatic potential of colorectal cancer cells (CRC) in the alcohol-injured liver. (a) The potential effects of alcohol on the direct processing of CEA by Kupffer Cells (KCs), the release of cytokines, and the role of those factors in facilitating the expression of pro-metastatic adhesion molecules on liver sinusoidal endothelial cells (LSECs). (b) The indirect processing of CEA by KCs and hepatocytes and the potential ethanol-elicited impairments to this process can enhance KC activation and pro-metastatic changes in the liver. Key: a-CEA asialo-CEA, ASGPR hepatic asialoglycoprotein receptor, CEA carcinoembryonic antigen, CEAR CEA receptor
17.4.1.1 Effect of Alcohol on Kupffer Cells
Kupffer cells are known to play an important role in ethanol and/or its metabolite’s detrimental effects on liver function. Therefore, the activation of liver macrophages and related consequences are of central importance. Kupffer cell (KC) activation results in the secretion of inflammatory cytokines and cytotoxic products including LPS-stimulated TNF-α production through signaling mediated by the KC macrophage endotoxin receptor, CD14. The evidence for KC involvement in ALD was confirmed when enhanced measures of injury (aminotransferase levels, steatosis, and cell death) were alleviated by gadolinium chloride-induced elimination of KCs in the liver [62]. In addition to ethanol-induced endotoxin-mediated activation of KCs and the consequential secretion of cytokines, KCs play multiple roles in the injured liver including cell adhesion, phagocytosis, and as a mediator of inflammation [63]. However, the contribution of liver macrophages in disease states is extremely complex due to the knowledge that activated KCs release different chemokines, reactive oxygen intermediates, and cytokines in response to ethanol or other stimuli. In support of our hypothesis that the effects of alcohol would facilitate enhanced CEA-mediated KC responses, it is important to note that the cytokine TNF-α has been linked to pro-metastatic changes of the microenvironment. It was demonstrated that TNF-α serum concentrations correlated to the increased risk of metastasis of certain tumors through the up-regulation of adhesion molecules on the endothelium [64]. In the setting of alcohol, the production of TNF-α is one of the earliest responses to damage implicating these events as key players in the pathogenesis of ethanol-induced liver injury [8, 65]. Notably, in recent studies we have determined that alcohol administration results in the enhanced KC production of TNF-α in response to CEA (Fig. 17.2). In this work, KCs were isolated from rats fed the Lieber–DeCarli control or ethanol-containing diets for up to 6 weeks, a well-established rodent model of alcohol-induced liver injury [66]. The obtained KCs were used in an in vitro assay in which they were stimulated in the presence of either human CEA or low concentrations of LPS as a positive control for KC stimulation, followed by analysis of the media for the presence of TNF-α. The results demonstrate that KCs from both control and ethanol-fed rats were stimulated to produce TNF-α in response to CEA as well as LPS as expected. Importantly, the KC response from alcohol-fed animals was found to be enhanced over that produced by KCs obtained from the pair-fed control animals. This data supports our hypothesis that the alcohol-mediated effects on liver macrophages contributes to increases in TNF-α expression, a cytokine known to potentiate metastatic processes.
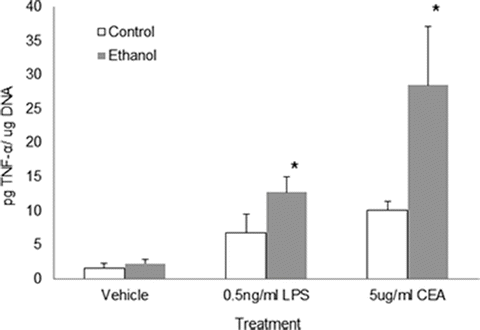
Fig. 17.2
Alcohol administration results in enhanced production of TNF-α from KCs in response to CEA. Kupffer cells (KCs) were isolated from the livers of rats chronically fed control or alcohol-containing liquid diet. TNF-α production was measured in the media of cultured KCs after 4 h of incubation with vehicle, LPS, or CEA as indicated. Values significant from control-fed animals are indicated (*p<0.05)
17.4.1.2 Alcohol-Mediated Defects to the Hepatocyte ASGPR and Asialo-CEA Processing
Alcohol consumption can lead to a variety of pathological consequences including alcohol-induced alterations to the essential resident cell of the liver parenchyma, the hepatocyte [67]. Studies by our Liver Study Group reported that particular processes are highly susceptible to the detrimental effects of ethanol as defects were identified in protein trafficking pathways, including the cellular process of RME [68–70]. Those works demonstrated that ethanol administration significantly impacted the function of the abundant hepatocyte ASGPR that was involved in the observed RME defects. It was shown that ethanol feeding to animals resulted in impairments to the ASGPR from initial binding of ligands and internalization of receptor–ligand complexes to its subsequent sorting and delivery to lysosomes for degradation [10, 11]. Several sites of alcohol-mediated alterations in the ASGP receptor were recorded that included a significant decrease in number of surface receptors and the ability to internalize and degrade ligands. The hepatocellular RME process is thought to be especially important in the regulation of CEA in the liver. In particular, the asialo-CEA that is produced by KCs can be recognized, bound to, and internalized via the ASGPR which facilitates the ultimate degradation of the CEA glycoprotein in the hepatocyte. In our current work, we examined how ethanol-mediated impairments to the ASGPR would affect the clearance of asialo-CEA by performing binding and degradation assays using radiolabeled desialylated CEA (asialo-CEA) and a known ligand for the ASGPR, asialoorosomucoid (ASOR) (Fig. 17.3). As predicted from our previous work, we found that ASOR binding to the surface of hepatocytes was impaired in cells from ethanol-fed animals compared to controls and that the binding was specific for ASGPR (inhibited by excess unlabeled ASOR). Importantly, we determined that asialo-CEA binding was impaired in hepatocytes from ethanol-fed animals and furthermore, was inhibited by unlabeled ASOR, demonstrating specificity to the ASGPR. Additionally, the terminal degradation of asialo-CEA in normal and ethanol-injured hepatocytes was measured (Fig. 17.4). Again, classic ethanol-induced reductions were observed in the degradation of ASOR, a well-characterized ligand for the ASGPR. In a similar fashion, the degradation of asialo-CEA was also found to be significantly reduced in hepatocytes obtained from chronically fed animals compared to controls. It is evident from these initial studies that ethanol-induced derangements of hepatocellular endocytosis can disrupt the regulation of CEA in the liver by altering effective clearance mechanisms.
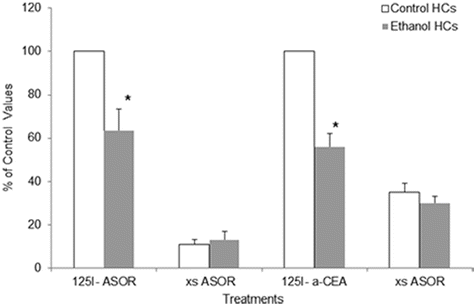
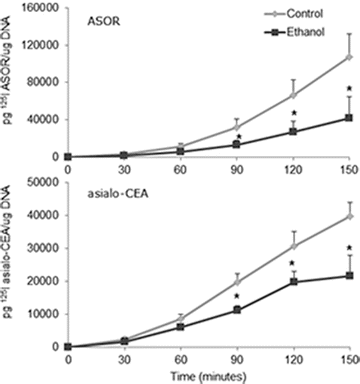
Fig. 17.3
Alcohol administration alters the binding of desialylated CEA to the hepatocyte asialoglycoprotein receptor (ASGPR). Hepatocytes (HCs) were isolated from control or ethanol-fed rats and incubated at 4 °C for 60 min with either radiolabeled asialoorosomucoid (125I ASOR) or asialo-CEA (125I a-CEA). Nonspecific binding was assessed by the inclusion of 100-fold excess of unlabeled ligand (xASOR). Values significant from control-fed HCs are indicated (*p<0.05)
Fig. 17.4
Degradation of asialo-CEA is impaired in hepatocytes isolated from ethanol-fed rats. Hepatocytes isolated from rats chronically fed control or alcohol-containing diets were incubated over a 150 min time course with radiolabeled desialylated CEA (125I asialo-CEA) or a specific ligand for the hepatocyte asialoglycoprotein receptor, asialoorosomucoid (125I ASOR). The degradation of the iodinated ligands was determined by the acid-soluble radioactivity measured in the cell culture media. Values significant from control-fed animals are indicated (*p<0.05)
17.5 Summary
Alcohol consumption has been shown to be a major etiologic factor of both acute and chronic diseases affecting many other critical organ systems (e.g., liver, pancreas, brain, and lung). In addition, it is clear that alcohol is a recognized carcinogen and that chronic and/or heavy consumption is a significant risk factor for the development of cancers as well as metastasis to secondary organs. Sadly, the development of liver metastasis in CRC patients has been correlated to alcohol consumption. Due to this correlation, alcohol consuming CRC patients require intensive examination and follow-up with respect to liver metastasis since most recurrences to the liver are not resectable leading to poor survival rates. Even though it is known that alcohol consumption correlates with liver metastasis and poor outcomes in CRC patients, the mechanism(s) by which alcohol participates in, or provides an environment supportive of CRC metastases is unknown. Some of the current research in this field is aimed at characterizing the role of CEA, a hallmark predictive factor of metastatic colon cancer. The relationships between serum CEA levels in alcoholics, ethanol-mediated alterations to liver cells and the consequences of alcohol-related CEA processing in the liver are being investigated. Understanding CEA processing in the liver has the potential to significantly impact the field and lead to the development of therapies for early detection and/or prevention of liver involvement in CRC disease.
References
1.
2.
3.
5.
Thomas P, Zamcheck N, Rogers AE, Fox JG (1982) Plasma clearance of carcinoembryonic antigen and asialo carcinoembryonic antigen by the liver of the nutritionally deficient rhesus monkey. Clin Lab Med 2:459–467PubMed
6.
7.
Thakur V, Pritchard MT, McMullen MR, Wang Q, Nagy LE (2006) Chronic ethanol feeding increases activation of NADPH oxidase by lipopolysaccharide in rat Kupffer cells: role of increased reactive oxygen in LPS-stimulated ERK1/2 activation and TNF-alpha production. J Leukoc Biol 79:1348–1356PubMedCentralPubMedCrossRef
8.
Thurman RG (1998) II. Alcoholic liver injury involves activation of Kupffer cells by endotoxin. Am J Physiol 275:G605–G611PubMed
9.
10.
Casey CA, Kragskow SL, Sorrell MF, Tuma DJ (1987) Chronic ethanol administration impairs the binding and endocytosis of asialo-orosomucoid in isolated hepatocytes. J Biol Chem 262:2704–2710PubMed
11.
Tworek BL, Tuma DJ, Casey CA (1996) Decreased binding of asialoglycoproteins to hepatocytes from ethanol-fed rats. Consequence of both impaired synthesis and inactivation of the asialoglycoprotein receptor. J Biol Chem 271:2531–2538PubMedCrossRef
Related posts:
 A Perspective on Chemoprevention by Resveratrol in Head and Neck Squamous Cell Carcinoma
Transgenic Mouse Models for Alcohol Metabolism, Toxicity, and Cancer
A Perspective on Chemoprevention by Resveratrol in Head and Neck Squamous Cell Carcinoma
Transgenic Mouse Models for Alcohol Metabolism, Toxicity, and Cancer
 Genetic Polymorphisms of Alcohol Dehydrogense-1B and Aldehyde Dehydrogenase-2, Alcohol Flushing, Mean Corpuscular Volume, and Aerodigestive Tract Neoplasia in Japanese Drinkers
Genetic Polymorphisms of Alcohol Dehydrogense-1B and Aldehyde Dehydrogenase-2, Alcohol Flushing, Mean Corpuscular Volume, and Aerodigestive Tract Neoplasia in Japanese Drinkers
 Alcohol Consumption, Wnt/β-Catenin Signaling, and Hepatocarcinogenesis
Alcohol Consumption, Wnt/β-Catenin Signaling, and Hepatocarcinogenesis
 Alcoholic Cirrhosis and Hepatocellular Carcinoma
Alcoholic Cirrhosis and Hepatocellular Carcinoma
 Synergistic Toxic Interactions Between CYP2E1, LPS/TNFα, and JNK/p38 MAP Kinase and Their Implications in Alcohol-Induced Liver Injury
Synergistic Toxic Interactions Between CYP2E1, LPS/TNFα, and JNK/p38 MAP Kinase and Their Implications in Alcohol-Induced Liver Injury
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


