Fig. 12.1
Alcohol and HCV in liver disease: common sites of action
12.2 Alcohol, HCV and Innate Immunity
The immune response is a common target of both alcohol and the HCV. HCV is a typical tissue-tropic virus that replicates in hepatocytes and triggers innate and adaptive immune responses [26, 27]. Recognition of HCV by innate immune cells such as monocytes and macrophages leads to inflammatory cytokine induction and type I IFN production [28–30]. Recognition of HCV by dendritic cells and their antigen presentation to T cells is pivotal in a robust anti-HCV immune response that leads to viral clearance. In effective resolution of acute HCV, DCs induce a strong CD4 Th1-type T cell activation that is HCV specific to lead to viral elimination. In contrast, during chronic HCV infection, DC functions are impaired and T cell activation is predominantly Th2 type and not antigen specific [31–33].
Importantly, alcohol also impairs immune responses. Acute alcohol binge results in attenuation of inflammatory cytokine induction and attenuation of Type I IFN induction [34–36]. Chronic alcohol equally impairs immunity and increases nonspecific inflammatory responses and decreases virus-induced Type I IFN production [35, 37].
These immune abnormalities in the local environment of alcohol exposed, HCV-infected liver can then contribute to a detrimental imbalance of innate immune responses that otherwise are critical in HCV elimination. Moreover, the chronic inflammatory signals and inflammatory cell presence and activation may promote HCC development.
12.2.1 Antiviral Immunity in HCV Infection and Alcohol Use
HCV is a single stranded RNA virus that is recognized by pattern recognition receptors (PRRs) on the host cells, that induce Type I IFNs and inflammatory mediators [29, 35]. In the HCV, several vital components represent “dangerous signals” for the host immune system (Fig. 12.2). The ssRNA of HCV is recognized by TLR8 while during HCV replication, the dsRNA is sensed by the host TLR3, RIG-I, and perhaps Mda-5. All of these PRRs induce type I IFNs, particularly IFNß in hepatocytes. In immune cells, HCV-induced activation of these PRRs can also trigger IFNα and IFNß (type I IFNs), IFN-gamma (γ) (type II), and IFN-lambdas (Type III IFNs) [38–40]. HCV, however, has several ways to undermine these host immune alert mechanisms. For example, the HCV NS3-4a serine protease cleaves the host adapter molecules that are critical in TLR3 and RIG-I activation, respectively [41, 42]. Current therapies with protease inhibitors target this activity and now are in clinical practice for treatment of HCV [43–45].
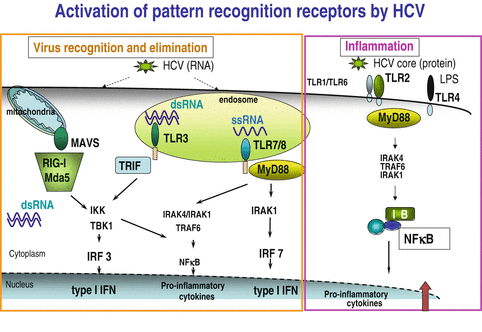
Fig. 12.2
Activation of pattern recognition receptors by HCV
Antiviral immunity is compromised by both acute and chronic alcohol use [46–49]. In a binge drinking model, blood mononuclear cells of human volunteers showed impaired type I IFN production in response to stimulation with viral danger signals via TLR8 or with bacterial danger molecule (LPS) that is recognized by TLR4 [35]. More important, similar defects were seen after chronic alcohol treatment of immune cells suggesting that alcohol, whether acute or chronic, impairs Type I IFN induction [35, 50, 51]. The combined negative effects of alcohol and HCV were also seen on IFN induction in hepatoma cells [52].
12.2.2 Mechanisms of Inflammation
HCV components, such as the core and NS3 proteins can be recognized by TLR2 and induce downstream activation of pro-inflammatory cascades in immune cells [53–56]. This has direct clinical relevance because in the HCV-infected liver there is increased infiltration and activity of innate immune cells including tissue macrophages and Kupffer cells that produce inflammatory mediators [57–60]. Even in peripheral blood monocytes that can serve as precursors of liver macrophages, there is increased in vivo activation and hyper-inflammatory response in patients with chronic HCV infection [60, 61]. It has been shown that the inflammatory cell and cytokine milieu contribute to triggering and promoting liver cirrhosis and cancerous transformation in hepatocytes [62, 63].
Chronic alcohol abuse is another trigger of inflammation. Activation of the inflammatory cascade, increased liver, and circulating pro-inflammatory cytokine levels are the characteristics of ALD and alcoholic steatohepatitis [64–66]. Several key elements have been identified in the pathomechanisms of ALD including LPS/TLR4-mediated activation of inflammation, upregulation of the inflammatory cytokine cascade (TNFα, IL-1ß, MCP-1, IL-6), that are linked to an in vivo Kupffer cell activation and sensitization to LPS. There is evidence for increased circulating levels of LPS, a gram negative bacterial innate immune danger signal in ALD in humans as well as in animal models [34, 67, 68]. Interestingly, serum levels of LPS are also increased in patients with treatment-naïve chronic HCV infection [60]. In vitro experiments demonstrated that chronic alcohol exposure augments TLR-induced TNFα production in human monocytes [35].
12.3 Cancer Surveillance in HCV Infection and Alcohol Use
12.3.1 NK Cells
Host immunity serves as a “controller” to prevent the development of cancer by recognizing and eliminating cells with cancerous malformation [69, 70]. However, alcohol is a major inhibitor of many of the key functions of the innate immune system that are pivotal in this process. For example, alcohol interferes with function of natural killer cells (NK cells) [71–73]. Deregulated activation of NK cells through a reduction of systemic β-endorphins production can also occur with alcohol consumption leading to increased hepatocyte damage [74]. Alternatively, recent reports have suggested a critical inhibitory role of alcohol on NK cells to effectively carry out immune surveillance. For example, alcohol consumption can suppress the effective function of NK cells in the liver by decreasing the expression of TRAIL, IFN-γ, and NKG2D—the activating NK cell receptor [75, 76]. Additionally, alcohol use blocks NK cell release from the bone marrow and significantly induces splenic NK cell apoptosis [77] which can compromise viral clearance and exacerbate disease progression during HCV infection. Finally, alcohol has also been shown to increase serum corticosterone levels which can impair the function of NK cells [78] and enhance HCV disease progression. Given the important role of NK cell in viral clearance, patients with a genetic predisposition for lower NK cell function usually progress to chronic HCV infection [79].
During HCV infection of hepatocytes, NK cells are rapidly activated providing an important role in the resolution of early infection [80, 81]. However, HCV infection can also interfere with NK cell activation and inhibit NK cell IFNγ production [82, 83]. Impaired activity of NK cells has been proposed as a mechanism contributing to HCV persistence. HCV chronic infection severely affects NK phenotype and function. It exhibits a polarized phenotype with increased cytotoxicity [84, 85]. In addition to their antiviral function, NK cells can suppress the development of fibrosis by directly killing activated myofibroblasts (MFB) and IFN-α production which induces cell cycle arrest and apoptosis of MFB [86, 87]. Chronic ethanol feeding diminished the inhibitory effect of NK/IFN-γ in liver fibrosis, which has been suggested to be an important mechanism contributing to alcohol acceleration of liver fibrosis in patients with chronic HCV infection [75, 87, 88].
12.3.2 Antigen Presenting Cells in HCV Infection
In addition to NK cells, antigen presenting cells are also important in recognition of injured and abnormal cells. However, both alcohol and HCV infection alone and together attenuate the capacity of myeloid dendritic cells to fulfill their antigen presenting and T cell stimulatory function [89–92]. Some of the mechanistic aspects of alcohol- and HCV-induced impairment of DC phenotype and function are similar and additive. For example, both alcohol and HCV infection results in impaired capacity of monocyte-derived DCs to reach full maturation after an external stimulation [91, 93]. This involves decreased expression of co-stimulatory molecules, decreased IL-12 and increased IL-10 production [91, 93]. In HCV infection, there is also overexpression of the PD1 ligand that is an inhibitory cell surface molecule for T cells [94–97]. Other types of the dendritic cell population are also negatively impacted in HCV infection. Plasmacytoid DCs, that are the major producers of IFNα, have reduced numbers in the periphery and most important, decreased capacity to produce IFNα [30, 98]. The mechanisms for this involve some of the inflammatory mediators produced by monocytes such as IL-10 and TNFα [30, 99, 100]. Most recently, IFN-lambda (IL-18 and IL-29) production was found in a unique DC population, the M2 DCs [40]. In HCV infection, however, this population has impaired production of IFN-lambda [40]. The biological and clinical consequence of this finding is under active investigation, however, one of the effects of IFN-lambda is to trigger and amplify production of other interferons [40]. Thus, decreased IFN-lambda in chronic HCV infection can have broad and magnified effects.
12.4 Alcohol and HCV Replication, Role of Micro-RNAs
12.4.1 Oxidative Stress
Despite the substantial epidemiologic data on HCV, alcohol and liver disease, the molecular mechanisms modulating chronic liver disease development and progress to fibrosis, cirrhosis, and even HCC are not fully known. Recent in vitro and mice studies expressing HCV proteins have been instrumental in deciphering some of the mechanisms underlying the synergism of alcohol abuse and HCV infection in liver disease [101–106]. The expression of the HCV core protein in mice caused significant production of reactive oxygen species (ROS) which appears to be responsible for mitochondrial DNA damage during HCV infection [105–108]. These observations clearly demonstrated a mechanistic insight by which the HCV core protein through synergistic significant oxidative stress induction can exacerbate liver injury in alcohol-fed core transgenic mice. Additionally, chronic alcohol use and HCV infection independently increase TLR2 and TLR4 expression in hepatocytes, Kupffer cells, and peripheral monocytes in both in vitro and in vivo studies [25, 35, 109–111]. Increased synergistic TLR expression during HCV infection and alcohol will mechanistically enhance TLR-mediated signal activations resulting in liver disease progression ultimately resulting in HCC [25, 110]. Mechanistic evidence has been demonstrated by chronic ethanol feeding in mice expressing the HCV NS5A protein in a hepatocyte-specific manner were mice fed with alcohol were prone to liver tumor development in a TLR4 dependent manner [10]. The tumorigenic effect of alcohol in a TLR4 dependent manner in hepatic HCV NS5A transgenic mice was also associated with the upregulation of the NANOG usually associated with HCC [112]. NANOG is transcription factor essential for self-renewal of stem cells [113, 114] and is associated with tumor malignancy and metastasis [115].
Chronic alcohol and HCV infection independently or in association also induce numerous biochemical and metabolic changes in hepatocytes that could directly or indirect initiate or potentiate HCC. Increased production of ROS and deposition of iron and their downstream effect have been advanced to at least in part account for the synergism of alcohol and HCV in mediating liver disease including HCC. The generation of ROS has been associated with both ALD and HCC. While there may be differences in their generation their downstream effects are quite similar. HCV core [116] and NS5A [117] have been shown to induce the generation of ROS via mitochondrial damage and calcium release. ROS also play an important role in alcohol-induced liver injury and in hepatocarcinogenesis [118]. In the liver, alcohol is metabolized by alcohol dehydrogenase and by cytochrome P450 2E1 (CYP2E1) to acetaldehyde—a carcinogen in mouse studies. Chronic alcohol use increases alcohol dehydrogenase and CYP2E1 which produces ROS in the presence or absence of alcohol. We and others have demonstrated the importance of CYP2E1 in enhancing HCV replication during alcohol exposure in CYP2E1-expressing hepatoma cells [119, 120]. Human studies on genetic polymorphisms alcohol dehydrogenase and CYP2E1 which metabolize alcohol in the development of HCC have provided conflicting findings. A Japanese study demonstrated an association between alcohol dehydrogenase and CYP2E1 gene polymorphism with the risk of HCC in humans while earlier a Korean study found no such association [121–123].
12.4.2 Modulation of HCV Replication by Alcohol Via HSP90
Alcohol abuse has been shown by numerous studies to exacerbate disease outcome during HCV infection. Recent studies have increasingly identified Heat-shock protein 90 (HSP90) as an important mediator of HCV disease progression associated with alcohol abuse. HSP90 is an evolutionary conserved chaperone protein that plays a key role with other co-chaperone proteins in assisting newly synthesized proteins to fold properly. They also assist in stabilizing proteins under stress and play a vital role in modulating protein degradation. HSP90 is normally induced during cellular stress conditions and numerous studies have demonstrated the diverse functions of HSP90 including those related to viral-pathogenesis, transcription regulation, neo-vascularization, and cancer development/metastasis [124–132]. It is estimated that over 16–20 % of cancers worldwide are caused by viruses [133, 134] including HCV. The molecular mechanisms by which HCV infection induces cancer development has been linked the expression viral oncogenes as well as the capacity for virus to induces sustained inflammation that promotes neoplastic transformation of infected cells [135]. While some oncogenic viruses have vaccines to prevent human infections, there is no vaccine against HCV. In this regard, it is therefore imperative to better understand the molecular mechanism by which HCV alone and in association with exacerbating factors like alcohol can promote cancer development. We and others have demonstrated that HSP90 increases during ALD, HCV infection, and even HCC development [136–141]. Mandrekar et al. recently demonstrated that alcohol-induced hepatic stress modulates alcohol liver disease in an HSP90 dependent manner [136, 138]. Additionally, we and others have demonstrated an important role of HSP90 modulating HCV replication with and without alcohol exposure [137, 142–146]. During the HCV life cycle, the HCV RNA is translated to form a single viral polyprotein. This HCV viral polyprotein is cleaved by cellular and viral proteins to form its nine functional viral proteins. HSP90 has been shown to play a critical role in the cleavage and activation of HCV NS3 and NS2 proteins [147]. The mechanism by which HSP90 induces such cleavage is yet to be identified but reports have speculated that indirect interaction between HSP90 and viral proteins might protect them from proteolysis [142, 147]. HSP90 also interacts with HCV NS5A protein and enhances viral replication in association with other host proteins [144, 148] which can promote cancer development.
In addition to its role in modulating HCV replication and viral protein stability, HSP90 has been shown to enhance the proliferative potential of malignant cells and even enable neoplastic cells to escape cell death ultimately enhancing neoplastic development [149, 150]. HSP90 also plays an important role in maintaining the integrity of NF-κB and Akt which are two main cell survival pathways that attenuate the anticancer efficacy of some cancer therapeutic drugs [151]. Inhibition of HSP90 has been shown to improve the efficacy of anticancer agents [152]. Given the important role played by HSP90 in ALD, HCV, and even HCC development, our findings and those by other groups show a great promise for HSP90 inhibitors as novel pharmacological agents for ALD, HCV, and anticancer therapy [136–138, 143, 153, 154]. Treatments against cancer targeting HSP90 are of prime importance and likely to be more successful since HSP90 is of great importance in maintaining the integrity, conformation, stability, and functional properties of important oncogenic proteins.
12.4.3 Role of Micro-RNA 122
Previous studies indicated that alcohol use can increase serum HCV levels [155, 156]; however, it remained unclear whether this was related to increased HCV replication. In a recent study, we demonstrated that alcohol increases HCV replication in vitro and identified a critical role of micro-RNA-122 (miRNA-122) in the process [119, 137] (Fig. 12.3), however a meta-analysis study showed that alcohol had no effect on HCV replication [157]. MiRNAs are non-coding RNAs that are increasingly being recognized as major players in the regulation of most physiologic and pathological processes. In the liver, miRNA-122 has the highest abundance in hepatocytes compared to other cell types [158, 159]. Furthermore, miRNA-122 is a host factor that has been shown to modulate the HCV replication machinery by binding to the HCV 5′-UTR [160–162]. Recently we discovered that alcohol increases miRNA-122 levels in hepatoma cells and through this mechanism, increases HCV replication [119]. Additionally, miRNA-122 regulation of HCV has been shown to be enhanced by the RISC-complex molecules Argonaute 2 (Ago2) [158] and GW182 which is increased during alcohol exposure in cultured human hepatic cell line Huh7.5 [137]. While RISC-complex proteins have been shown to enhance HCV replication in association with miRNA-122, a recent report indicates that Ago2 might be dispensable when miRNA-122 is overexpressed [163]. Interestingly miRNA-122 expression is decreased in the liver in HCC [164, 165]. In a mouse model of ALD, we found that chronic alcohol feeding in mice results in decreased miRNA-122 levels in the liver but increased levels in the serum [166]. Because serum miRNAs are highly stable, they have been proposed as potential biomarkers. Indeed, increased levels of miRNA-122 appear to correlate with the extent of liver damage and serum ALT levels in different mouse models of liver disease [166–170]. MiRNA-122 also regulates genes involved in lipid metabolism and in cell cycle. Recent findings have described the phenotype of miRNA-122 loss in hepatic lipid modulation [171, 172]. These findings demonstrated that miRNA-122 loss was associated with modulation of multiple hepatic pathways involved in fat metabolism, tumor suppression, inflammation, and even hepatic fibrosis. While loss of miRNA-122 in these studies induces steatohepatitis, it is unknown whether exogenous miRNA-122 might ameliorate fatty liver disease induced by diet or virus infection. Additionally, the tumor-suppressive function of miRNA-122 illustrated by both groups suggests that the restoration of miRNA-122 expression may be of benefit against HCC and even enhance response to therapy as recently proposed [173]. While this might prove beneficial for some HCC patients, such applications might need to carefully assess findings from current trails using miRNA-122 inhibitors to treat HCV infection [174, 175]. In addition, several other miRNAs are also dysregulated in HCC that may represent biomarkers or therapeutic targets (Table 12.1).
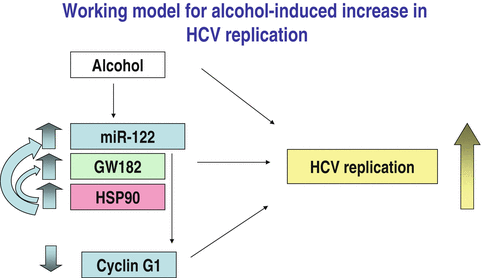
Fig. 12.3
Working model for alcohol-induced increase in HCV replication
Table 12.1
Characteristic micro-RNAs in HCC
Liver reduced levels of: | Serum reduced levels of: |
|---|---|
miR-24a | miR-21 |
miR-26a | miR-16 |
miR-15a/b | miR-199a |
miR-150 | miR-122 |
miR-195 | miR-22 |
miR-122 | |
miR-20 family | |
miR-124 | |
let-7 family members |
12.5 Summary
The current literature suggests that alcohol and HCV have several common targets in modeling liver disease (Fig. 12.4). Both affect and disable key immune functions particularly in innate immunity in antiviral host defense as well as in inflammation. Additional common element on the effect of increased gut permeability and the effect in modulating HCV associated with alcohol abuse have yet to be fully explored [176, 177]. Finally, in hepatocytes, the capacity of alcohol to modulate the cell cycle and the HCV replication process contributing to the development of HCC at the molecular level still needs to be addressed. Given the recent developments with new and potent drugs in the treatment of HCV infection [178–182], it is possible that the negative trend in liver cancer development in alcoholic-HCV patients may be significantly reduced.
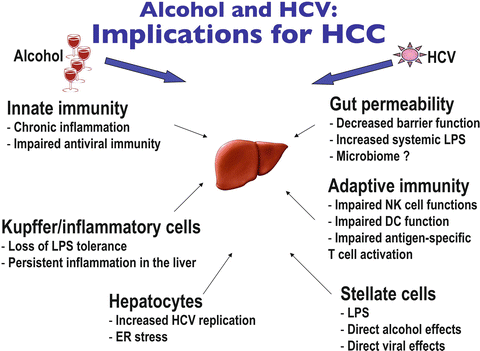
Fig. 12.4
Alcohol and HCV: implications for HCC
Conflict of Interest
The authors declare there are no conflicts of interest.
Grant Acknowledgment 5R37AA014372-10
References
1.
Davis GL, Dempster J, Meler JD, Orr DW, Walberg MW, Brown B, Berger BD, O’connor JK, Goldstein RM (2008) Hepatocellular carcinoma: management of an increasingly common problem. Proc (Bayl Univ Med Cent) 21:266–280
2.
Morgan TR, Mandayam S, Jamal MM (2004) Alcohol and hepatocellular carcinoma. Gastroenterology 127:S87–S96PubMed
3.
Sanyal AJ, Yoon SK, Lencioni R (2010) The etiology of hepatocellular carcinoma and consequences for treatment. Oncologist 15(Suppl 4):14–22PubMed
4.
Tornesello ML, Buonaguro L, Tatangelo F, Botti G, Izzo F, Buonaguro FM (2013) Mutations in TP53, CTNNB1 and PIK3CA genes in hepatocellular carcinoma associated with hepatitis B and hepatitis C virus infections. Genomics 102:74–83PubMed
5.
Levrero M (2006) Viral hepatitis and liver cancer: the case of hepatitis C. Oncogene 25:3834–3847PubMed
6.
Bartenschlager R, Penin F, Lohmann V, Andre P (2011) Assembly of infectious hepatitis C virus particles. Trends Microbiol 19:95–103PubMed
7.
O’Shea RS, Dasarathy S, McCullough AJ, Practice Guideline Committee of the American Association for the Study of Liver Diseases, Practice Parameters Committee of the American College of Gastroenterology (2010) Alcoholic liver disease. Hepatology 51:307–328PubMed
8.
Frazier TH, Stocker AM, Kershner NA, Marsano LS, McClain CJ (2011) Treatment of alcoholic liver disease. Therap Adv Gastroenterol 4:63–81PubMedCentralPubMed
9.
Szabo G, Lippai D (2012) Molecular hepatic carcinogenesis: impact of inflammation. Dig Dis 30:243–248PubMed
10.
Machida K (2010) TLRs, alcohol, HCV, and tumorigenesis. Gastroenterol Res Pract 2010:518674PubMedCentralPubMed
11.
Koike K, Tsutsumi T, Miyoshi H, Shinzawa S, Shintani Y, Fujie H, Yotsuyanagi H, Moriya K (2008) Molecular basis for the synergy between alcohol and hepatitis C virus in hepatocarcinogenesis. J Gastroenterol Hepatol 23(Suppl 1):S87–S91PubMed
12.
Rosman AS, Waraich A, Galvin K, Casiano J, Paronetto F, Lieber CS (1996) Alcoholism is associated with hepatitis C but not hepatitis B in an urban population. Am J Gastroenterol 91:498–505PubMed
13.
Schiff ER (1999) The alcoholic patient with hepatitis C virus infection. Am J Med 107:95S–99SPubMed
14.
Tanaka T, Yabusako T, Yamashita T, Kondo K, Nishiguchi S, Kuroki T, Monna T (2000) Contribution of hepatitis C virus to the progression of alcoholic liver disease. Alcohol Clin Exp Res 24:112S–116SPubMed
16.
Day CP (2001) Heavy drinking greatly increases the risk of cirrhosis in patients with HCV hepatitis. Gut 49:750–751PubMedCentralPubMed
17.
Neumann AU, Lam NP, Dahari H, Gretch DR, Wiley TE, Layden TJ, Perelson AS (1998) Hepatitis C viral dynamics in vivo and the antiviral efficacy of interferon-alpha therapy. Science 282:103–107PubMed
18.
Stickel F, Schuppan D, Hahn EG, Seitz HK (2002) Cocarcinogenic effects of alcohol in hepatocarcinogenesis. Gut 51:132–139PubMedCentralPubMed
19.
Enomoto N, Ikejima K, Yamashina S, Hirose M, Shimizu H, Kitamura T, Takei Y, Sato And N, Thurman RG (2001) Kupffer cell sensitization by alcohol involves increased permeability to gut-derived endotoxin. Alcohol Clin Exp Res 25:51S–54SPubMed
20.
Garcia-Tsao G, Wiest R (2004) Gut microflora in the pathogenesis of the complications of cirrhosis. Best Pract Res Clin Gastroenterol 18:353–372PubMed
21.
Wigg AJ, Roberts-Thomson IC, Dymock RB, McCarthy PJ, Grose RH, Cummins AG (2001) The role of small intestinal bacterial overgrowth, intestinal permeability, endotoxaemia, and tumour necrosis factor alpha in the pathogenesis of non-alcoholic steatohepatitis. Gut 48:206–211PubMedCentralPubMed
22.
Purohit V, Bode JC, Bode C, Brenner DA, Choudhry MA, Hamilton F, Kang YJ, Keshavarzian A, Rao R, Sartor RB, Swanson C, Turner JR (2008) Alcohol, intestinal bacterial growth, intestinal permeability to endotoxin, and medical consequences: summary of a symposium. Alcohol 42:349–361PubMedCentralPubMed
23.
Szabo G, Bala S (2010) Alcoholic liver disease and the gut-liver axis. World J Gastroenterol 16:1321–1329PubMedCentralPubMed
24.
Petrasek J, Mandrekar P, Szabo G (2010) Toll-like receptors in the pathogenesis of alcoholic liver disease. Gastroenterol Res Pract 2010:pii: 710381
25.
Szabo G, Aloman C, Polyak SJ, Weinman SA, Wands J, Zakhari S (2006) Hepatitis C infection and alcohol use: a dangerous mix for the liver and antiviral immunity. Alcohol Clin Exp Res 30:709–719PubMed
26.
Protzer U, Maini MK, Knolle PA (2012) Living in the liver: hepatic infections. Nat Rev Immunol 12:201–213PubMed
27.
Horner SM, Gale M Jr (2013) Regulation of hepatic innate immunity by hepatitis C virus. Nat Med 19:879–888PubMed
28.
Heydtmann M (2009) Macrophages in hepatitis B and hepatitis C virus infections. J Virol 83:2796–2802PubMedCentralPubMed
29.
Liu BS, Janssen HL, Boonstra A (2012) Type I and III interferons enhance IL-10R expression on human monocytes and macrophages, resulting in IL-10-mediated suppression of TLR-induced IL-12. Eur J Immunol 42:2431–2440PubMed
30.
Dolganiuc A, Chang S, Kodys K, Mandrekar P, Bakis G, Cormier M, Szabo G (2006) Hepatitis C virus (HCV) core protein-induced, monocyte-mediated mechanisms of reduced IFN-alpha and plasmacytoid dendritic cell loss in chronic HCV infection. J Immunol 177:6758–6768PubMed
31.
Fan Z, Huang XL, Kalinski P, Young S, Rinaldo CR Jr (2007) Dendritic cell function during chronic hepatitis C virus and human immunodeficiency virus type 1 infection. Clin Vaccine Immunol 14:1127–1137PubMedCentralPubMed
Related posts:
 A Perspective on Chemoprevention by Resveratrol in Head and Neck Squamous Cell Carcinoma
Transgenic Mouse Models for Alcohol Metabolism, Toxicity, and Cancer
A Perspective on Chemoprevention by Resveratrol in Head and Neck Squamous Cell Carcinoma
Transgenic Mouse Models for Alcohol Metabolism, Toxicity, and Cancer
 Genetic Polymorphisms of Alcohol Dehydrogense-1B and Aldehyde Dehydrogenase-2, Alcohol Flushing, Mean Corpuscular Volume, and Aerodigestive Tract Neoplasia in Japanese Drinkers
Genetic Polymorphisms of Alcohol Dehydrogense-1B and Aldehyde Dehydrogenase-2, Alcohol Flushing, Mean Corpuscular Volume, and Aerodigestive Tract Neoplasia in Japanese Drinkers
 Alcohol Consumption, Wnt/β-Catenin Signaling, and Hepatocarcinogenesis
Alcohol Consumption, Wnt/β-Catenin Signaling, and Hepatocarcinogenesis
 Alcoholic Cirrhosis and Hepatocellular Carcinoma
Alcoholic Cirrhosis and Hepatocellular Carcinoma
 Synergistic Toxic Interactions Between CYP2E1, LPS/TNFα, and JNK/p38 MAP Kinase and Their Implications in Alcohol-Induced Liver Injury
Synergistic Toxic Interactions Between CYP2E1, LPS/TNFα, and JNK/p38 MAP Kinase and Their Implications in Alcohol-Induced Liver Injury
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


