Carcinogenesis
1. Acetaldehyde
2. Oxidative stress: induction of cytochrome P450 2E1 and reactive oxygen species
3. Retinoids
4. Epigenetic changes
5. Cancer genes and signaling pathways
Mitogenic signals: MAPK, RAS
Insensitivity to antigrowth signals: Rb and cell cycle control, TGFβ
Apoptosis: p53, PTEN
Angiogenesis: VEGF
Metastasis: cell adhesion, ECM, osteopontin
Developmental signaling pathways
6. Inflammation
7. Immunosuppression
8. Organ-specific mechanisms: cirrhosis, gastroesophageal reflux
Disease, estrogens
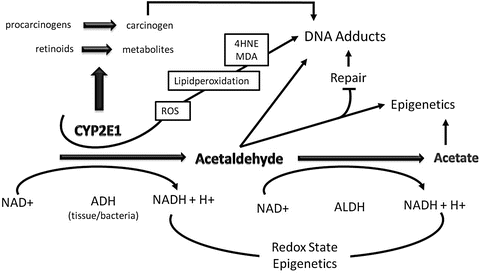
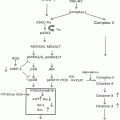
Fig. 4.1
The role of cytochrome P450 2E1 (CYP2E1) and acetaldehyde in ethanol-mediated carcinogenesis. Ethanol is metabolized via alcohol dehydrogenase (ADH) and CYP2E1 to acetaldehyde, and acetaldehyde is further metabolized via acetaldehyde dehydrogenase (ALDH) to acetate. Through the ADH and ALDH reaction reducing equivalence in the form of NADH are generated influencing the redox state of cell with major influence of epigenetics. Acetaldehyde by itself forms carcinogenic DNA adducts, inhibits DNA repair, and leads to epigenetic changes due to a decreased availability of methyl groups resulting in hypomethylation of DNA and histones. CYP2E1 is important in the metabolism of retinoids to their polar metabolites and of procarcinogens to their ultimate carcinogens. The loss of retinol and retinoic acids leads to dedifferentiation and hyperproliferation. In addition, ethanol metabolism via CYP2E1 results also in the generation of reactive oxygen species (ROS) with lipid peroxidation and the occurrence of lipid peroxidation products such 4-hydroxynonenal (4HNE) and malondialdehyde (MDA) which form highly carcinogenic exocyclic etheno DNA adducts. Finally, acetate may influence epigenetics due to hyperacetylation of histones
4.2.1 Acetaldehyde as a Carcinogen
4.2.1.1 Mechanisms
AA produced by various alcohol dehydrogenases (ADHs) is a toxin and carcinogen which rapidly binds to proteins and DNA. It is degradated by acetaldehyde dehydrogenases (ALDHs) to acetate, which is not toxic. AA has three major modes of action with respect to carcinogenesis:
1.
It forms with DNA carcinogenic DNA adducts.
2.
It inhibits DNA repair.
3.
It has significant effects on epigenetics (DNA methylation).
Ad (1) AA forms DNA adducts leading to N2-ethyl-2′-deoxyguanosine (N2-Et-dG) which is rather a marker for chronic alcohol consumption than a carcinogenic lesion. On the other hand, data by Dr. Brooks show clearly that AA under basic conditions and in the presence of basic amino acids, histones and polyamines, generates 1,N2-propano-2′-deoxyguanosine (PDG) which is a carcinogenic lesion [10]. This will be discussed by Dr. Brooks in a separate article of this book.
Ad (2) AA inhibits DNA repair and has an inhibitory effect on 3N-A-DNA-G and on MMST-DNA-G HOGG1. In addition, AA binds to glutathione and inhibits, therefore, the antioxidative defense system (AODS) which is responsible for the detoxification of reactive oxygen species (ROS) and reactive nitrogen species (RNS). Furthermore, AA stimulates NFκB which inhibits apoptosis, a major feature of carcinogenesis. AA can either act directly or via interleukin 6 (Il6) or MCL2 [11, 12].
Ad (3) Methylation of DNA and histones is an important mechanism in epigenetics. Alcohol interferes with methyl transfer at various sites. First of all, ethanol leads to a decreased intake of folic acid [13]. Folic acid is an important factor in the methylation of homocysteine to become methionine. Deficiency in folic acid leads to an accumulation on homocysteine and to diminished generation of methionine. Furthermore, AA inhibits methyl adenosine transferase 1 (MAT 1) with the result of a reduced production of S-adenosyl methionine (SAMe), the final precursor of methyl transfer. Subsequently alcohol also inhibits DNA methyltransferase. As a side effect alcohol also decreases vitamin B6 which is involved in these processes. In conclusion, there is a diminished production of SAMe with a diminished methyl transfer and diminished methylation of DNA and histones associated with an accumulation of homocysteine, an important component to enhance endoplasmic reticulum stress [14]. In this context it has to be pointed out that in addition to DNA and histone hypomethylation due to the mechanisms discussed, a hyperacetylation of histones also occurs due to two factors: (1) an increased production of acetate following ethanol oxidation and (2) a change in the hepatic redox state with an increase in NADH and a decrease in NAD again due to ethanol oxidation. These are prerequisites to inhibit the activity of histone deacetylase (HDAC) [15].
4.2.1.2 Genetic Aspects of Acetaldehyde Accumulation
Genetic linkage studies with alcoholics have provided strong support for the assumption that AA plays a central role in alcohol-associated carcinogenesis. These studies found that individuals who accumulate AA because they carry certain alleles of the genes encoding ADH or ALDH have an increased cancer risk [16, 17]. For both the ADH1B and the ADH1C genes, several alleles exist that result in differences in the activity of the ADH molecules that they encode. For example, the ADH1B*2 allele encodes an enzyme that is approximately 40 times more active than the enzyme encoded by the ADH1B*1 allele. Similarly, the enzyme encoded by the ADH1C*1 allele is 2.5 times more active than the enzyme encoded by the ADH1C*2 allele [18]. Individuals who carry the highly active ADH1B*2 allele rapidly convert ethanol to AA. This leads to AA accumulation following alcohol consumption and results in toxic side effects, such as a flushing syndrome with sweating, accelerated heart rate, nausea, and vomiting. These adverse symptoms exert a protective effect against acute and chronic alcohol consumption. The ADH1B*2 allele is rarely found in Caucasians but occurs more frequently in Asian populations. It has recently been demonstrated that also the low activity ADH1B*1/1* genotype associates with enhanced exposure to AA through saliva. This might be due to lower systemic elimination rate of ethanol from the body which results in prolonged exposure to acetaldehyde produced by oral microbes [19]. There is also strong evidence that the combination of ALDH2 deficiency and slow ADH1B associates with the highest risk for esophageal cancer especially among heavy drinkers [20, 21]. The effects of the different ADH1C alleles on alcohol metabolism and, consequently, on drinking levels and alcohol-related carcinogenesis are more subtle. They can be studied best in Caucasian populations in which the highly active ADH1B*2 allele is rare. Studies on the relationship between ADH1C alleles and cancer occurrence in Caucasians have led to contradictory results which have been discussed elsewhere [22]. Harty and colleagues [23] were the first who compared the risk of oral cancer associated with various alcohol consumption levels in individuals homozygous for the more active ADH1C*1 allele with the risk in heterozygotes who carried only one copy of this allele or were homozygous for the less active ADH1C*2 allele. The study found that individuals who consumed eight or more drinks per day and were homozygous for the more active ADH1C*1 allele had a 40-fold increased risk for oral cancer compared with nondrinkers. In contrast, people who consumed the same amount of alcohol but who were heterozygous or homozygous for the less active ADH1C* allele had only a four- to sevenfold increased risk compared with nondrinkers.
We determined ADH1C polymorphisms in more than 400 heavy drinkers with daily alcohol intake of more than 60 g and various cancers of the upper aerodigestive tract, liver, and breast [24–27]. Cases of cancer patients were compared with carefully matched control patients with alcohol-related diseases (e.g., cirrhosis of the liver, pancreatitis, and alcohol dependence) but no cancer. Cancer patients and control subjects were of similar age and had similar histories of alcohol consumption and cigarette smoking. In this study, significantly more patients with alcohol-related cancers either had at least one ADH1C*1 allele or were homozygous for ADH1C*1 than did patients with other alcohol-related diseases. Statistical analyses determined a significant association between ADH1C*1 allele frequency and rate of homozygosity and an increased risk for alcohol-related cancer (P < 0.001). Finally, individuals homozygous for ADH1C*1 had a relative risk of developing esophageal, liver, and head and neck cancers of 2.9, 3.6, as well as 2.2, respectively, compared with people homozygous for ADH1C*2. We also found that individuals who are homozygous for the ADH1C*1 allele had significantly higher AA levels in their saliva than did heterozygous individuals or individuals who are homozygous for the ADH1C*2 allele [24]. Saliva rinses the mucosa of the upper aerodigestive tract, and any AA in the saliva may be taken up by mucosal cells. Moreover, mucosal cells display little ALDH2 enzyme activity and therefore cannot efficiently detoxify AA. As a result, AA may bind to proteins and DNA in the mucosal cells and may initiate carcinogenesis. The hypothesis that AA in the saliva contributes to tumor development is supported by the observation that AA-fed rats with intact salivary glands showed excessive proliferation of the upper gastrointestinal mucosa [28], similar to the changes observed following chronic alcohol consumption [29]. When the glands were surgically removed (i.e., when the animals no longer produced saliva), however, this excessive cell proliferation disappeared [30].
In conclusion, the evidence for the involvement for AA in ethanol-associated cancer can be summarized as follows:
1.
High AA levels occur in the saliva and in the colon following alcohol consumption.
2.
AA is a toxin which binds to proteins leading to structural and functional alterations of the cell with epigenetic (disturbed DNA methylation and DNA reparation) and genetic effects. AA forms mutagenic and carcinogenic DNA adducts especially in hyperregenerative tissues.
3.
Elevated AA levels in experimental carcinogenesis accelerate cancer development in the colon.
4.
Data in humans demonstrate high risk for upper aerodigestive tract and colorectal cancer in individuals who accumulate AA due to decrease detoxification (ALDH2 deficiency) or increased production (ADH1c1 homozygosity).
4.2.2 Alcohol and Oxidative Stress
4.2.2.1 Mechanisms of ROS and Cytochrome P450 2E1 in Alcohol-Mediated Carcinogenesis
Various factors contributed to oxidative stress in alcoholic liver disease (ALD) including inflammation and the invasion of neutrophils and macrophages. However, one major factor is the induction of cytochrome P450 2E1 (CYP2E1) after chronic alcohol consumption. This induction varies interindividually and occurs already at an alcohol dose of 40 g per day as well as already at a time interval of one week [31]. As longer as alcohol is consumed as higher is the induction. However, it is important to note that some individuals have no CYP2E1 induction at all. It is believed that individuals who have a strongly induced CYP2E1 status have an increased risk for ALD due to the production of ROS and their consequences. After alcohol withdrawal CYP2E1 disappears rapidly within days [31]. Approximately 10 to 15 % of alcohol oxidation occurs via CYP2E1. However, in the induced state, more than 30 % may be metabolized via CYP2E1. In this situation ROS occurs. ROS may lead to lipid peroxidation with a generation of lipid peroxidation products such as malondialdehyde and 4-hydroxynonenal (4HNE) [11].
Furthermore, CYP2E1 is involved in the activation of various xenobiotics and procarcinogens to their ultimate carcinogenic intermediates which is important in situations where carcinogens are consumed together with alcohol [11]. For example, aflatoxins occur in certain food products contaminated with fungi. These aflatoxins are activated through CYP2E1 which is induced after alcohol consumption [32]. CYP2E1 is also involved in drug metabolism including paracetamol and isoniazid. In this context it is referred to other review articles [33].
Finally, CYP2E1 catalyzes the metabolism of retinol and retinoid acid (RA) leading to a decrease in RA concentrations associated with loss of cell differentiation and cellular hyperregeneration [34]. These low levels in RA lead to a decrease of the RXR and RAR receptor as well as to a decrease in MKP-1 and an increase of JNK. At the same time CYP2E1 leads to oxidative stress which by itself increases JNK leading to an increased phosphorylation of c-fos and c-jun, the proteins of AP1 [35]. Both increased AP1 expression and decreased RXR and RAR receptor regulate cell proliferation and apoptosis and lead to an increased cellular proliferation which stimulates carcinogenesis. On the other hand, if CYP2E1 is inhibited by CMZ, oxidative stress decreases, RA increases, and carcinogenesis is prevented [36]. It has been shown that the supplementation of vitamin A or RA is not helpful in preventing carcinogenesis since both compounds in the presence of an induced CYP2E1 are metabolized to polar retinoic metabolites which have apoptotic properties [37].
4-HNE is well known to form exocyclic ethanol DNA adducts with DNA which are highly carcinogenic. It has been shown in CYP2E1 knockout mice that carcinogenic DNA lesions occur more frequently [38]. In recent experiements we could show that in CYP2E1 overexpressing HepG2 cells an increased exocyclic ethanol adduct formation with increasing concentrations of alcohol exists an increased exocyclic ethanol adduct formation with increasing concentrations of alcohol [39]. The occurrence of these adducts could be diminished significantly in the presence of clomethiazole (CMZ), a specific CYP2E1 inhibitor. These cell experiments could be extended to the human situation [39]. In biopsies from patients with ALD a significant correlation between CYP2E1, 4-HNE and exocyclic ethanol adducts of adenine and cytosine was found. In a recent nonpublished study, we have examined 89 patients with various stages of non-cirrhotic ALD, and we clearly found that CYP2E1 was not associated with the amount of alcohol consumed which is not surprising, keeping in mind the fact that the induction of CYP2E1 is interindividually different. However, there was a significant correlation between etheno-DNA adduct formation and CYP2E1 induction (Seitz, personal communication).
Related posts:
 Transgenic Mouse Models for Alcohol Metabolism, Toxicity, and Cancer
Transgenic Mouse Models for Alcohol Metabolism, Toxicity, and Cancer
 Alcohol and Breast Cancer: Reconciling Epidemiological and Molecular Data
Alcohol and Breast Cancer: Reconciling Epidemiological and Molecular Data
 Alcohol Consumption, Wnt/β-Catenin Signaling, and Hepatocarcinogenesis
Alcohol Consumption, Wnt/β-Catenin Signaling, and Hepatocarcinogenesis
 Fetal Alcohol Exposure Increases Susceptibility to Carcinogenesis and Promotes Tumor Progression in Prostate Gland
Fetal Alcohol Exposure Increases Susceptibility to Carcinogenesis and Promotes Tumor Progression in Prostate Gland
 Alcoholic Cirrhosis and Hepatocellular Carcinoma
Alcoholic Cirrhosis and Hepatocellular Carcinoma
 Synergistic Toxic Interactions Between CYP2E1, LPS/TNFα, and JNK/p38 MAP Kinase and Their Implications in Alcohol-Induced Liver Injury
Synergistic Toxic Interactions Between CYP2E1, LPS/TNFα, and JNK/p38 MAP Kinase and Their Implications in Alcohol-Induced Liver Injury
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


