Fig. 2.1
Consumption of 20 % alcohol enhanced tumorigenesis in mice. Ovariectomy abolished this effect despite alcohol-induced increase in estrogen levels (From Wong et al. [61])
The results of this study highlight the importance of assessing the HER2 status in addition to that of ER and PR. To translate these results to humans, women who take estrogen-containing HRT could have an increase in breast cancer risk due to the combined effects of HRT and alcohol. However, the epidemiological study (#26 above) that took into account the HER2 status found that in premenopausal women with ER+/PR+/HER2+ tumors, the risk of developing breast cancer for those drinking 5 to ≤23 g/day was increased by 61 % and for ER−/PR−/HER2+ by 92 % for women drinking the same amount. It is apparent that the results of this epidemiological study do not dovetail in a straightforward manner with the mouse study, suggesting that the relationships between these variables are more complex.
The use of HRT that contains estrogen adds to the complexity of the interaction between various risk factors. For example, the Women’s Health Initiative (WHI) reported that women who received ≥5 years of continuous treatment with estrogen and progestin have increased risk of breast cancer [62]. Similar results were reported by studies #12, #13, and #15 above, but not by #4. In the same WHI study, postmenopausal women with prior hysterectomy who received estrogen alone showed a statistically significant decrease in breast cancer risk [63]. In addition, women in the French observational E3N study who received estrogen alone or estrogens combined with micronized progesterone showed no increase in breast cancer risk; however, those who received estrogens and androgenic progestins, or who were on HRT for long time, were at increased risk [64]. In a Finnish study [65], postmenopausal women using estradiol (E2)-progestogen therapy showed no increase in breast cancer incidence within the first 3 years of use.
Since supraphysiological estrogen doses caused mammary adenocarcinomas in rats [66], and alcohol consumption increased plasma estrogen levels (not to a supraphysiological level) in human female volunteers [67], it was postulated that alcohol use should be more strongly associated with ER+ than ER− tumors. However, epidemiological studies that assessed the risk of alcohol consumption based on tumor status were contradictory. For example, while some epidemiological studies showed a modest increase in ER+ tumors with the consumption of 15–30 g/day, there was no association with consumption of >30 g/day (see study #8 above). The link between alcohol use and ER+ or PR+ status was not consistent across different studies (see discussion under #13). For instance, studies showed statistically nonsignificant associations with either or both ER+, PR+ for women consuming ≥10 g/day (study #2). Consumption of 30 g/day was associated with a modest increase in risk of ER+/PR+ tumors, but there was no increase in risk for ER+/PR− tumors (study #12). Other studies showed that 10 g/day of alcohol increased the risk in either ER+ or ER− (study #13), or mostly in ER−/PR− (study #14). Study #17 showed that consumption of 10–20 g/day was associated with 7 and 28 % increase in risk for ER+/PR+ and ER−/PR− tumors, respectively. Finally, meta-analysis of 4 prospective and 16 case–control studies [68] showed that an increase in alcohol consumption of 10 g per day was associated with increased risks for ER+/PR+ (11 %) and ER+/PR− (15 %). The authors concluded that the observed positive associations with alcohol for ER+/PR+ and ER+/PR− tumors cannot be explained by estrogen-dependent pathway only.
In addition, estrogen status is influenced by numerous exogenous factors. For instance, persistent exposure of mammary gland stem and progenitor cells to different environmental factors such as xenoestrogens (bisphenol A, phthalates, ethinyl estradiol, phytoestrogens) alters their epigenetic reprogramming during epithelial differentiation [69]. This is mediated, in part, through ERα nuclear receptors which activate or silence the transcription of target genes [70]. Interactions between ERα and various enzymes involved in histone modifications (histone acetyltransferases, histone deacetylases, histone methyltransferases, histone demethylases), co-activators, and co-repressors have introduced another layer of complexity in the epigenetic regulation of breast carcinogenesis [71]. Furthermore, women who were exposed in utero to diethylstilbestrol (DES), a synthetic estrogen, are at greater risk of developing breast cancer in their 40s (1.8–2.5-fold increased risk) and in their 50s (threefold increased risk) [72]. These environmental and epigenetic factors involving estrogen need to be taken into consideration in epidemiological studies.
Estrogen levels are intertwined with obesity to influence breast cancer risk. While some studies showed no effect of BMI on risk for breast cancer (e.g., study #20 above), dysregulation of sex hormones, hyperinsulinemia, and inflammatory cytokines in obese women are factors that could influence the risk for breast cancer. Obesity is significantly associated with low plasma levels of sex-hormone-binding globulin (SHBG), which increases the bioavailability of estrogens and androgens [73]. Thus, many established risk factors for breast cancer may function through an endocrine mechanism.
2.4.2.2 Alcohol Metabolism
The liver is the major organ for metabolizing ethanol mainly by oxidative pathway which involves cytosolic alcohol dehydrogenase (ADH), of which multiple isoenzymes exist—e.g., in humans, class I ADH is composed of three genes (ADH1A, ADH1B, and ADH1C)—to produce acetaldehyde, a highly reactive molecule. ADH acts on a wide range of substrates including retinol. The cytochrome P450 isozymes, mainly CYP2E1, predominantly present in the endoplasmic reticulum, also contribute to ethanol oxidation to acetaldehyde in the liver, particularly at higher alcohol concentrations. CYP2E1-dependent ethanol oxidation may occur in other tissues where ADH activity is low. CYP2E1 also produces highly reactive oxygen species (ROS), including hydroxyethyl, superoxide anion, and hydroxyl radicals. Acetaldehyde, produced by ethanol oxidation through any of these mechanisms, is rapidly metabolized mainly by mitochondrial aldehyde dehydrogenase (ALDH2) to form acetate and NADH and to a much lesser extent by ALDH1 in the cytosol. Mitochondrial NADH is oxidized by the mitochondrial electron transport chain. Chronic alcohol consumption renders mitochondrial oxidative phosphorylation inefficient by interfering with the main respiratory complexes (Complex I, III, IV, and V) of the electron transport system encoded on mitochondrial DNA (mtDNA), resulting in the formation of the superoxide anion. In breast cancer, like in other cancers, mitochondrial function is severely impaired [74]. One early event in breast carcinogenesis can be mutations in mtDNA that destabilize the oxidative phosphorylation system (OXPHOS) which can result in a shift in energy metabolism toward enhanced aerobic glycolysis. Alcohol metabolism could influence breast carcinogenesis by generating acetaldehyde and ROS and by interfering with retinol metabolism.
Acetaldehyde is suspected in playing a role in breast carcinogenesis. Since blood acetaldehyde levels are very low or undetectable after alcohol consumption in humans [75], the human breast would not be exposed to significant levels of exogenous acetaldehyde. Thus, ADH activity and in situ generation of acetaldehyde in the human breast tissue after ethanol consumption is of potential significance. In mammary tissue of rats, cytosolic ADH and ALDH1 activities were 5.8 and 8.3 % of that in the liver of the same animals, respectively [76]. Similarly, mitochondrial ALDH2 activity in breast tissue was 7.1 % of that in the liver. In humans, studies on normal and neoplastic breast tissue showed that class I, but not class IV, ADH is expressed in human mammary epithelium, which can support ADH-mediated oxidation of ethanol; however, the expression of class I ADH is dramatically reduced or abrogated in invasive breast cancers [77]. The authors opined that this “virtual abrogation of expression of class I ADH in invasive breast cancer suggests that the enzyme has some ‘tumor suppressor’ function in the mammary epithelium.” However, whether the reduction in class I ADH activity was causally related to tumor formation or was merely a bystander effect was not considered. To investigate acetaldehyde formation by the cytosolic pathway and the microsomal fraction in the mammary tissue, Sprague–Dawley female rats were injected intraperitoneally with 0.8 mL ethanol/kg/day for four consecutive days [78]. Mammary microsomal metabolism of alcohol to acetaldehyde by CYP2E1 was not induced after ethanol (or acetone) treatment, despite reports that CYP2E1 is expressed in normal and cancerous breast tissue [79]. In contrast, the cytosolic fraction of alcohol treated animals showed higher concentrations of acetaldehyde.
In humans, the interaction between alcohol consumption and ADH2 polymorphism with respect to breast cancer risk was reported in 278 German women with invasive breast cancer [80]. The authors stated that breast cancer risk associated with alcohol consumption may vary according to ADH2 polymorphism, probably due to differences in alcohol metabolism.
Variations in ADH and ALDH activities were reported to influence the risk of breast cancer. For example, while the ADH1B genotype [81] was not associated with breast cancer risk in a German population, a role for the ADH1C genotype has been suggested. This genotype, which is expressed mainly in the liver but also in breast tissue, has three polymorphic genes: ADH1C1*1 and ADH1C1*2 genotypes which result in enzymes with fast and intermediate turnover rates and which increase the risk of breast cancer in Chinese drinkers, compared to ADH1C2*2 which results in an enzyme with a slow rate of metabolism [82]. Similar results were obtained in a Long Island Breast Cancer Study which genotyped 1,047 breast cancer cases. Consumption of 15–30 g/day was associated with OR of 2.0, 1.5, and 1.3 in ADH1C1*1, ADH1C1*2, and ADH1C2*2 genotypes, respectively [83]. Ironically, another study in Caucasian postmenopausal women found an association between risk of breast cancer and the slow metabolizing variant, which led the authors to conclude that “ethanol rather than acetaldehyde is related to breast cancer risk” [84]. However, two studies found no association between breast cancer risk and functional allelic variants of the ADH1B and 1C genes [85] and ADH1B and ALDH2. The authors concluded that “our findings do not support the hypothesis that acetaldehyde is the main contributor to the carcinogenesis of alcohol-induced breast cancer” [86].
Acetaldehyde and NADH produced by alcohol metabolism can be substrates for xanthine oxidoreductase (XOR), which is inducible by alcohol and produces ROS, especially superoxide anion [87]. To add to the complexity, XOR also metabolizes (activates) nitrofurans and nitroimidazoles, chemicals that are used in veterinary medicine and by beekeepers in honey-producing hives. Therefore, residues of these compounds could exist in animal-derived foods and honey and might be involved in the associated mammary carcinogenic effects [88].
To explain acetaldehyde’s role in carcinogenesis, scientists proposed a model in which acetaldehyde reacts with DNA to generate DNA lesions that form interstrand cross-links (ICLs). Cells are protected against replication blocking DNA lesions and ICLs through the Fanconi anemia-BRCA (FANC-BRCA) DNA-damage response network. Mutations in two major susceptibility genes, BRCA1 and BRCA2, which are involved in the maintenance of genomic integrity and DNA repair, were identified as major risk factors for breast cancer [89, 90]. The role of high levels of acetaldehyde in activating the FANC-BRCA network was discussed elsewhere [91, 92]. Furthermore, polymorphisms in the DNA repair gene XRCC1 was associated with increased breast cancer risk in African-American women [93].
The role of acetaldehyde in breast carcinogenesis has not been evident in epidemiological studies. For example, study #19 above did not find a significant association between breast cancer risk and alcohol consumption in Asian American women, almost half of whom are deficient in ALDH2, the mitochondrial enzyme that metabolizes acetaldehyde. Furthermore, study #20 reported that the increase in risk for breast cancer in Japanese population was not modified by alcohol-induced facial flushing, which means the risk was not modified in women who have the defective ALDH2*2.
To examine alcohol effects on oxidative stress in the mammary tissue, female Sprague–Dawley rats were fed alcohol for 28 days [78]. An increase in hydroperoxide, but not the lipid peroxidation product malondialdehyde (MDA) concentration, and a significant reduction in glutathione in mammary tissue were observed. A study by Li and colleagues comparing breast cancer patients with cancer-free women reported that the levels of hydroxyl radical-DNA adducts and MDA-DNA adducts were ninefold higher in patient’s normal breast tissue adjacent to tumor tissue than in breast tissue from cancer-free controls [94]. These reports highlight the potential that oxidative stress may lead to DNA damage in cancer patients that is not evident in healthy women.
Class I ADH has the potential to catalyze the oxidation of retinol (vitamin A) to retinal [95], the first step in the biosynthesis of retinoic acid (RA), the principal mediator for maintaining epithelia in a differentiated state. Chronic and excessive alcohol intake interferes with retinoid metabolism and results in reduced RA. Alcohol acts as a competitive inhibitor of oxidation of vitamin A to RA (which involves ADH and ALDH) and induces CYP2E1, which can enhance catabolism of vitamin A and RA. The biological activity of RA is primarily mediated by nuclear retinoid receptors which are involved in the antitumor activity of retinoids. Studies indicate cross-talks between classic retinoids and various intracellular pathways controlling the growth, survival, and invasive/metastatic behavior of breast cancer cells [96]. Impaired RA homeostasis interferes with signaling (e.g., downregulates retinoid target gene expression) and with “crosstalk” with the mitogen-activated protein kinase signaling pathway (MAPK), including Jun N-terminal kinase and p38 kinase [97]. These observations could have implications for breast cancer prevention. However, better understanding of the alcohol–retinoid interaction and the molecular mechanisms involved is needed before it would be justified to pursue retinoids in the prevention of breast cancer. Nonetheless, retinoids could be potential components of innovative and rational therapeutic combinations for breast cancer. Yet, it is important to evaluate the responsiveness of ER+ tumors to retinoids and whether HER2 expression always plays a negative role in modulating retinoid sensitivity of HER2+/ER+ mammary tumors, as suggested by some studies [98].
2.4.2.3 Folate Metabolism/Epigenetic Factors
Mutations in oncogenes and tumor suppressor genes result in specific gene expression profiles that are involved in the regulation of cellular homeostasis, including cell proliferation and DNA repair and survival. However, differentiation of mammary stem cells to primitive progenitor cells is under epigenetic control. Epigenetic mechanisms, which result in changes in gene expression patterns without altering DNA sequence, partake in mammary glands developmental phases from in utero to menopause, as well as in breast carcinogenesis. One of these epigenetic mechanisms is DNA methylation [99].
DNA methylation involves the transfer of a methyl group from S-adenosylmethionine (SAM)—by DNA methyltransferases (DNMTs)—onto the 5′-position of the cytosine residue found in cytosine guanosine dinucleotide pairs (CpG). SAM is generated from methionine. After the methyl transfer reaction, SAM forms S-adenosylhomocysteine, which is then broken down to homocysteine. The latter can be remethylated to form methionine, by transferring a methyl group either from N5-methyltetrahydrofolate (THF) by methionine synthase or from betaine by betaine-homocysteine methyl transferase. Hypermethylation of CpG groups renders affected loci inaccessible to transcription factors resulting in transcriptional silencing. Importantly, CpG methylation in promoter regions of tumor-suppressor genes (e.g., BRCA1) leads to the inactivation of these cancer-preventing proteins. Similarly, hypermethylation of numerous genes, whose biological function include hormone regulation, DNA repair, cell cycle regulation, tissue remodeling, apoptosis, cell adhesion and invasion, cell growth inhibition, and angiogenesis, has been identified in breast tumors [100]. Furthermore, DNA hypermethylation results in aberrant regulation of the Wnt pathway in breast cancer [101]; BRCA1 expression is suppressed by a combination of gene mutation and DNA hypermethylation [102]. One epidemiological study of the interactions between alcohol consumption and breast cancer risk in BRCA1 and BRCA2 mutation carriers reported no significant interaction with BRCA1 mutations but a greater risk of alcohol-associated breast cancer in women with BRCA2 mutations [103]. In fact, the same investigators reported an inverse association between breast cancer and current alcohol consumption in women with a BRCA1 mutation [104]. Another study reported no association between alcohol intake and breast cancer risk for women with BRCA1 or BRCA2 mutations and suggested a possible reduction in risk in BRCA2 mutation carriers with “modest” alcohol intake [105].
DNA hypomethylation can also contribute to breast carcinogenesis [106]. For example, promoter hypomethylation could reactivate the expression of certain proto-oncogenes (such as synuclein γ) which are associated with tumor metastasis [107].
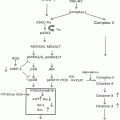
Chronic heavy alcohol consumption leads to substantial DNA hypomethylation as a result of significant reduction in tissue SAM (Fig. 2.2). Additionally, alcohol perturbs the folate cycle that is involved in the methionine metabolic pathway, which is integral to supplying the methyl groups necessary for DNA methylation. Folate is an important nutrient required for DNA synthesis, and at least 30 different enzymes are involved in the complex folate cycle including methylenetetrahydrofolate reductase (MTHFR), methionine synthase (MTR), and methionine synthase reductase (MTRR). Defects or polymorphic variations in the folate metabolic pathway may influence cancer susceptibility. However, a study on 1,063 women with breast cancer found no association between MTHFR genotype and risk for breast cancer, and there was no evidence of an interaction of genotype and alcohol consumption in premenopausal women. However, in postmenopausal women, there was an increase in breast cancer risk in those who were homozygote TT for MTHFR C677T and drank >1.9 drinks/day [108].
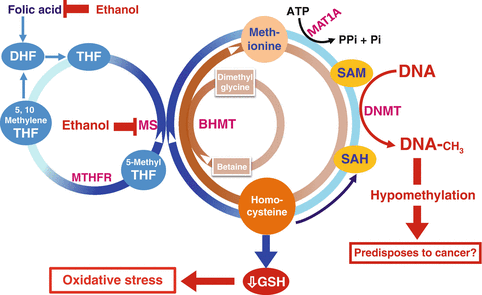
Fig. 2.2
Alcohol’s effects on homocysteine/methionine metabolism and DNA methylation. MTHFR methylene tetrahydrofolate reductase, MAT methionine adenosyltransferase, HCC hepatocellular carcinoma, BHMT betaine homocysteine methyltransferases, GSH glutathione, ATP adenosine triphosphate, Pi inorganic phosphate
Chronic heavy alcohol consumption can cause relative folate deficiency due to the negative effects of alcohol on folate metabolism, including malabsorption, increased excretion, or enzymatic suppression. Based on the above interactions between folate and alcohol, it would be expected that high folate intake should ameliorate the association between alcohol intake and risk for breast cancer that is caused by this mechanism. Examination of epidemiological studies revealed inconsistencies in the findings. For example, studies discussed under #25 and #26 showed that folate intake attenuated the alcohol-associated risk for breast cancer, so did another epidemiological study in Anglo-Australian [109] women aged 40–69 years; whereas studies discussed under #20 and #23 showed no association. Other studies reported that high intake of folic acid increased the risk of breast cancer in postmenopausal women enrolled in the Prostate, Lung, Colorectal, and Ovarian Cancer Screening Trial [110] cohort in the USA. Additionally, a case–control study in 570 Thai women concluded that genetic polymorphisms in folate and alcohol metabolic pathways may contribute to the etiology of breast cancer among Thai women [111]. In conclusion, the impact of folate supplementation on the risk for alcohol-induced breast cancer probably is affected by a wide range of other factors that are not well understood.
2.5 Concluding Remarks
Breast cancer is the most diagnosed cancer in women worldwide; it is one of the primary causes of death among women globally. Women, particularly if they have known genetic susceptibilities, should consult with their physician about risk factors involved in breast cancer. Overconsumption of alcohol is a risk factor for many diseases and one that women with or without a high risk for breast cancer are well advised to avoid,
The question whether a woman should not drink at all in order to reduce the risk of breast cancer or may drink moderately without undue risk is not settled. The reasons are threefold: (1) there are at least 20 recognized risk factors that can affect the onset and outcome of breast cancer (Fig. 2.3) and the overall risk depends on the interactions of all these factors, including those that have not yet been identified; (2) epidemiological studies resulted in contradictory associations between the amount of alcohol consumed and risk for breast cancer; and (3) discrepancies exist between epidemiological and molecular studies.
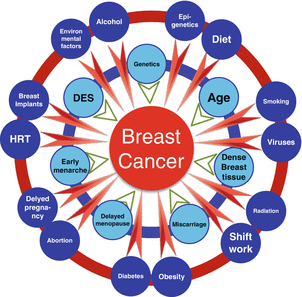
Fig. 2.3
Multiple risk factors associated with breast cancer
2.5.1 Multiple Risk Factors
Numerous risk factors are involved in breast carcinogenesis; only a few of them will be discussed below to illustrate the complex interactions between these risk factors. Some of the risk factors associated with breast cancer are genetic and beyond the control of a woman. Mutations in BRCA1 and BRCA2, were identified as major risk factors for breast cancer [89, 90]. However, the incomplete penetrance of these mutations suggests that other factors, environmental and hormonal, may modify that risk, for example, a study in which monozygotic (MZ) twins who carried identical BRCA1 gene mutation resulted in discordant phenotypes; one suffered from breast cancer twice in 27 years while her MZ twin remained healthy [112].
The majority of breast cancers are not hereditary. Most late-onset breast cancer occurs in the absence of a first-degree family history of breast cancer. Genome-wide association studies have identified genetic susceptibility variants of medium-penetrance [113–116] (which confer risk of 2–3-fold per allele) and modest penetrance (which increase the risk 1.1–1.3-fold per allele). However, these variants could explain only 20–25 % of familial breast cancer risk [117]. These findings led to the hypothesis that susceptibility to breast cancer is polygenic, i.e., conferred by a large number of loci, each with limited contribution to breast cancer risk [118].
Age is another important nonmodifiable risk factor. Invasive breast cancer or its precursor lesion DCIS occurs at an exponential rate until about age 50 (menopause) followed by a slower rate of increase [119], supporting the notion that breast cancer biology is age-dependent. Early-onset breast cancer, therefore, could largely represent inherited mutations (BRCA1, BRCA2, TP53, ATM, or PTEN) or early life transforming events that affect the immature mammary cells [120]. In contrast, late-onset breast cancer could be due to an early mutagenic initiating event, which is then subjected to later life exposure to endogenous or exogenous promoting agent(s) and further compounded by age-related impairments in macromolecular repair, immune surveillance, or xenobiotic detoxification. This could explain the increased risk from HRT.
Risk factors are greatly influenced by epigenetic mechanisms that change gene expression patterns for cell differentiation, proliferation, and apoptosis. These epigenetic mechanisms include DNA methylation, histone modifications, and the effects of noncoding RNAs such as microRNAs (miRNA) [99]. Epigenetic changes that influence the transciptomes, proteomes, and metabolomes and ultimately breast cancer evolution, are brought about by numerous endogenous (e.g., hormonal, microbiota, aging, inflammation, inherited diseases such as type 1 diabetes) and exogenous (diet, smoking, infection, alcohol, obesity, radiation, shift work, circadian rhythm disturbances) etiological factors that result in the vast heterogeneity of breast cancers. In fact, a study on the clinically relevant subtypes, luminal A and basal-like breast cancer revealed that distinct molecular mechanisms might have been preprogrammed at an early stage of the disease and that these breast tumor subtypes represent biologically distinct disease entities that may require different therapeutic strategies [121].
The interactions between genes and the environment are crucial. Environmental factors add another layer of complexity by their interactions with the susceptibility genes for breast cancer and for metabolic diseases. For example, in a twin study in Finland [122], the probability that a co-twin would develop breast cancer (given that one twin already had breast cancer) was 10 % for monozygotic and 8 % for dizygotic twins, suggesting that combined environmental effects are dominant in the development of breast cancer. In addition, in a subset of the Million Women Study (study #11 above), the strongest suggestion of a gene–environment interaction was between the high-risk common variant CASP8-rs1045485 and alcohol consumption; the per-allele relative risk of breast cancer for CASP8-rs1045485 was not increased by consuming <1 drink/day, but was increased by 23 % (nonsignificant) in women who reported consuming one or more alcoholic drinks/day [123]. Another study found no interaction between the breast cancer susceptibility locus CASP8-rs17468277 and consuming <20 g/day of alcohol; however risk was increased by 45 % in those who drank ≥20 g/day [124]. Therefore, epidemiological studies should focus on gene–environment interactions rather than singling out individual risk factors as if they operate in isolation.
2.5.2 Epidemiological Studies
While epidemiological studies, when conducted properly, can be effective tools to identify the root of health problems and disease outbreaks in a community, epidemiological studies that deal with alcohol consumption and its consequences should be interpreted with considerable caution. Such studies can only highlight associations and cannot determine cause-and-effect relationships. Considering the wide range of variations in the size of studies, measurement errors in input and outcome variables, and individual variations in genetic background and life style factors, many of which were not taken into account, it is not surprising that the results have been contradictory. The epidemiological studies on alcohol and breast cancer discussed above point to the following important points as possible sources of the variability in outcomes:
1.
All studies use self-report to determine the amount and type of alcoholic beverage consumed. Most of the studies acknowledged “recall bias” that makes the alcohol consumption variable notoriously inaccurate, and drinking pattern could change during the period under consideration. In fact, a recent study on moderate alcohol intake and cancer stated that the apparent increased risk of cancer among light-moderate drinkers may be “substantially due to underreporting of intake” [125]. There is now strong evidence that recall over prolonged periods tends to grossly underestimate actual drinking frequency. This was highlighted dramatically in a recent study by Stockwell et al. [126] who compared Quantity-Frequency (QF) recall with beverage-specific “yesterday” (BSY) consumption reports and with alcoholic beverage sales reports in a Canadian population to demonstrate that underreporting of alcohol consumption was considerable (2–3-fold) and varied by age and consumption level. Particularly relevant is the finding that moderate drinkers underreport their drinking much more than more frequent drinkers. In many of the epidemiological studies summarized above, an underreporting by a factor of 2–3 would increase the alcohol consumption associated with increased risk of breast cancer into the recognized risky drinking category. Other studies used as input of alcohol consumption “ever” vs. “never” without even quantification, and others use inaccurate information to characterize drink size (e.g., study #1 characterizes a drink of a liquor as one ounce containing 15.1 g of alcohol). The definition of a standard drink according to the National Institute on Alcohol Abuse and Alcoholism [127] and the Dietary Guidelines for Americans is: 12 oz of beer (5 % alcohol), 5 oz of wine (12 % alcohol), and 1.5 oz of distilled spirit (40 % alcohol). In addition, the Dietary Guidelines for Americans define moderate drinking as consuming no more than one drink/day for a woman, and no more than 2 drinks/day for a man. Although some studies used FFQ and claim it as valid, lack of consumption ascertainment can result in confusing results. For example, in study #4 above, consumption of 1 or 2 drinks/day increased breast cancer risk by 40 %, whereas consumption of two or more drinks/day resulted in no increase in risk. A 42 % decrease in breast cancer risk was associated with drinking <1.5 drink/day (study #5), whereas drinking 3–6 drinks/week was associated with a 15 % increase in risk (study #2). In fact, a meta-analysis reported by Longnecker (#3) stated “the modest size of the association and variation in results across studies leave the causal role of alcohol in question.”
2.
Patterns of consumption are rarely analyzed. The majority of studies use average weekly consumption. Drinking seven drinks on a Saturday night and nothing the rest of the week is not, health-wise, the same as having one drink every day of the week.
3.
Breast cancer develops over a relatively long period of time, often more than 20 years [128], and thus, correlations between alcohol consumption and breast cancer cannot be determined in epidemiological studies with windows of alcohol exposure that captures current or recent alcohol intake, after clinical diagnosis; for example, studies # 4, 6, 14, and 21 assessed recent alcohol consumption, consumption 2 years prior to diagnosis, past-year drinking, or consumption 3 months after diagnosis, respectively.
4.
Despite the fact that age per se is a risk factor for breast cancer and during aging other environmental factors may compound or modify the risk, most epidemiological studies stratify data based on pre- or postmenopausal status alone. Thus, including women (ages 20–74, 25–85, 40–75, 23–74, and 20–87 in studies # 4, 5, 7,16, and 21, respectively)—who have various degrees of exposure to environmental and lifestyle factors that influence the genesis of breast cancer—without stratification could be confounding. Similarly, studies that took into account other risk factors such as smoking, BMI, use of HRT, etc., resulted in contradictory findings (see the above cited studies).
5.
 A Perspective on Chemoprevention by Resveratrol in Head and Neck Squamous Cell Carcinoma
Transgenic Mouse Models for Alcohol Metabolism, Toxicity, and Cancer
A Perspective on Chemoprevention by Resveratrol in Head and Neck Squamous Cell Carcinoma
Transgenic Mouse Models for Alcohol Metabolism, Toxicity, and Cancer
 Alcohol Consumption, Wnt/β-Catenin Signaling, and Hepatocarcinogenesis
Alcohol Consumption, Wnt/β-Catenin Signaling, and Hepatocarcinogenesis
 Fetal Alcohol Exposure Increases Susceptibility to Carcinogenesis and Promotes Tumor Progression in Prostate Gland
Fetal Alcohol Exposure Increases Susceptibility to Carcinogenesis and Promotes Tumor Progression in Prostate Gland
 Alcoholic Cirrhosis and Hepatocellular Carcinoma
Alcoholic Cirrhosis and Hepatocellular Carcinoma
 Synergistic Toxic Interactions Between CYP2E1, LPS/TNFα, and JNK/p38 MAP Kinase and Their Implications in Alcohol-Induced Liver Injury
Synergistic Toxic Interactions Between CYP2E1, LPS/TNFα, and JNK/p38 MAP Kinase and Their Implications in Alcohol-Induced Liver Injury




Results of the association between breast cancer risk and alcohol consumption based on ER, PR, and HER2 status are very contradictory and vary between different studies. Therefore, correlations with HRT use cannot be deciphered, based on currently available data.
Related posts:
A Perspective on Chemoprevention by Resveratrol in Head and Neck Squamous Cell Carcinoma
Transgenic Mouse Models for Alcohol Metabolism, Toxicity, and Cancer
Alcohol Consumption, Wnt/β-Catenin Signaling, and Hepatocarcinogenesis
Fetal Alcohol Exposure Increases Susceptibility to Carcinogenesis and Promotes Tumor Progression in Prostate Gland
Alcoholic Cirrhosis and Hepatocellular Carcinoma
Synergistic Toxic Interactions Between CYP2E1, LPS/TNFα, and JNK/p38 MAP Kinase and Their Implications in Alcohol-Induced Liver Injury
Stay updated, free articles. Join our Telegram channel
Full access? Get Clinical Tree
