Fig. 1
Cell signalling pathways implicated in hepatocarcinogenesis
Overexpression of HGF and c-MET occurs in 33 and 20–48 % of human HCC tumors, respectively [41–45] and appears to be associated with earlier stage tumors with favorable pathologic features [46]. The prognostic value of c-MET overexpression is controversial; while some investigators have found no relation with survival outcomes [46], others have found that overexpression portends a poor prognosis [45, 47].
c-MET receptor antagonist is currently being assessed in several clinical trials. ARQ 197, a selective inhibitor of the c-MET RTK, is the subject of an ongoing randomized, placebo-controlled phase II study [48]. Foretinib (GSK 1363089), a dual inhibitor of c-MET and VEGFR2, is also being studied in a phase I/II study (www.clinicaltrials.gov, NCT00920192).
1.2 Angiogenesis Inhibition
Angiogenesis is a vital process for tumor growth and survival. The highly vascular nature of HCC arises due to the influence of several important mediators of angiogenesis including angiopoietins [49], vascular endothelial growth factor (VEGF), platelet-derived growth factor (PDGF; [50]), fibroblast growth factor (FGF), and their respective receptors [51]. Targeting of the angiogenesis pathway has proven to be a highly successful tactic in the management of HCC, offering the possibility of longer term survival and disease control.
1.2.1 Anti-VEGF Antibodies
Inhibition of VEGF is one of the best studied approaches to arresting angiogenesis, and has been shown to provide meaningful clinical benefits in a growing array of cancers. Derangements in VEGFR signaling may occur through VEGF ligand overexpression, receptor alterations resulting in its constitutive activation [51], in response to environmental stressors like hypoxia [52], or through interactions with other growth factor-related signaling cascades such as the EGFR and their downstream effectors [51, 53]). VEGF/VEGFR activity appears to be implicated in hepatocarcinogenesis, as VEGFR-3 is upregulated in the presence of hepatitis B x antigen [52], and VEGF overexpression has been found in cirrhotic and dysplastic liver tissue [53]. Furthermore, VEGF and VEGFR overexpression in the serum and tissue of patients with established HCC relative to surrounding normal liver are associated with a more aggressive phenotype [56–58] and a poorer prognosis [59–61]. Compared to metastatic tumors which replace the hepatic parenchyma, HCC lesions tend to displace the parenchyma and rely on vascular outgrowth to survive. This is reflected on imaging where HCC lesions appear hypervascular, unlike hepatic metastases [62]. All of these findings support the VEGF pathway as a vital target in the management of HCC.
The anti-VEGF antibody, bevacizumab, has shown activity in HCC. A phase II trial of bevacizumab monotherapy at two different doses reported a disease control rate of 67 %, including partial responses in 12.5 % of the 24 evaluable patients. Decreases in the levels of circulating endothelial cells were associated with responses [63]. Major dose-limiting toxicities occurred in 20 % of patients, including variceal hemorrhage, transient ischemic attacks, hemorrhagic ascites and, proteinuria. Another phase II study of bevacizumab monotherapy documented an objective response rate of 13 %, 6-month progression-free-survival rate of 65 %, a median progression-free-survival time of 6.9 months, and 1-, 2-, and 3-year survival rates of 53, 28, and 23 %, respectively [64]. Major grade 3/4 toxicities occurred in 4–15 % of patients and included hypertension, arterial and venous thrombotic events, and one fatal hemorrhagic event due to esophageal varices. The inclusion criteria were subsequently modified, mandating that varices be identified and treated before protocol enrollment [65]. The real-time effects of bevacizumab were manifested by decreases in tumor enhancement on dynamic contrast-enhanced MRI (DCE-MRI), as well as decreases in serum VEGF-A, and stromal-derived factor-1 levels. Outcomes did not differ between dose levels [64].
Combination of bevacizumab with chemotherapy doublets, while tolerable, does not appear to be more efficacious than bevacizumab alone. A phase II study of bevacizumab with gemcitabine and oxaliplatin yielded partial responses in 20 % of patients, stable disease in 27 %, a 6-month progression-free-survival rate of 48 %, and median progression-free and overall survival times of 5.3 and 9.6 months, respectively. Grade 3/4 toxicities included fatigue, hypertension, and myelosuppression [66]. Another phase II study combining bevacizumab with capecitabine and oxaliplatin produced a similar partial response rate of 11 %, stable disease in 78 %, a 6-month progression-free-survival rate of 40 %, and a median progression-free survival time of 5.4 months [67]. Bevacizumab-related toxicities included one case of gastrointestinal perforation and sepsis, and two variceal bleeds.
In contrast to the rather modest results with chemotherapy, a phase II trial of bevacizumab plus erlotinib produced more striking progression-free survival and overall survival times of 9 and 15.7 months, respectively [68]. Progression-free survival at 16 weeks was 62.5 %, and the disease control rate was 68 %, including 10 partial responses. The majority of patients had an ECOG <1, and 87.5 % had Child-Pugh A cirrhosis.
1.2.2 Multitargeted Receptor Tyrosine Kinase Inhibitors
Sorafenib is an inhibitor of Raf and VEGFR-2 and -3, PDGFR-βc-k it and FLT-3 receptor tyrosine kinases [69]. The clinical potential of sorafenib in HCC was first recognized following a phase II trial in 137 treatment-naïve patients with an ECOG performance status of 0–1, 72 % of whom had Child-Pugh A cirrhosis [70]. Although the study reported a near negligible response rate with partial and minor responses in only 2.2 and 5.8 % of patients, respectively, 34 % of patients achieved disease stability lasting at least 16 weeks. The median time to progression and overall survival times were 4.2 and 9.2 months, respectively. Major grade 3–4 toxicities were fatigue, diarrhea, and hand-foot syndrome which occurred in 5.1–9.5 % of patients. No significant pharmacokinetic differences were seen between hepatitis B and C infected patients. Pretreatment immunohistochemical staining intensity for phosphorylated extracellular signal regulated kinase (pERK), a downstream effector of Raf (see section on targeting intracellular signaling cascades), correlated with time-to-progression. An 18-gene expression panel derived from blood cell RNA distinguished between responders and nonresponders. Another key finding was that a subset of responding patients actually appeared to display tumor growth in association with increased central tumor necrosis during the arterial phase of triphasic liver CT scans. Calculation of the ratio of tumor necrosis to tumor volume using a computer algorithm [71] was shown to be associated with disease control [72]. The use of this parameter in HCC response is currently being prospectively validated.
The survival benefit with sorafenib was subsequently established in two multicenter randomized placebo-controlled phase III trials. The Sorafenib HCC Assessment Randomized Protocol Trial (SHARP) trial enrolled a total of 602 patients, 82–83 % of whom had advanced (BCLC stage C) disease [73]. Dominant causes of cirrhosis were HCV infection in 27–29 %, alcohol in 26 %, HBV infection in 18–19 %. and other/unknown in 25–29 %, most likely representing non-alcoholic steatohepatitis and other metabolic disorders. Liver function was preserved with 95–98 % of patients having Child-Pugh A cirrhosis. Despite a partial response rate of only 2 %, significant prolongations in overall survival (10.7 versus 7.9 months, p<0.001) and time to radiographic progression (5.5 versus 2.8 months, p<0.001) were observed with sorafenib. Dose-limiting toxicities were hand-foot syndrome and rash or desquamation, and diarrhea. Grade 3–4 bleeding events were rare (<1 %).
The other phase III trial was conducted in an Asian patient population consisting of 271 patients, 94–97 % of whom had Child-Pugh A cirrhosis and an ECOG of 0–1 [74]. In contrast to the SHARP trial, over 70 % of patients were infected with HCV and >95 % had Barcelona Clinic Liver Cancer (BCLC) stage C disease. The median overall survival (6.5 versus 4.2 months, p = 0.014) and TTP (2.8 versus 1.4 months, p = 0.0005) were in favor of sorafenib. The toxicity profile was also similar to the findings of the SHARP trial. Objective responses were seen in only 1.3–3.3 % of patients.
It is notable that the magnitude of benefit with sorafenib observed in the study by Cheng et al. though significant, was smaller than in the SHARP trial. This difference has been attributed to the greater proportion of patients with advanced disease in the Asian study (>95 versus 82–83 % on SHARP; [75] as well as the differential sensitivities to sorafenib conferred by HCV compared to HBV infection. Several investigators have observed that patients infected with HCV survive longer with sorafenib compared to those with other causes of liver disease [76–78]. In particular, a retrospective subset analysis of the initial phase II sorafenib study revealed that patients infected with HCV tended to survive longer than those infected with HBV [78]. These findings might be explained by HCV core protein-induced upregulation of the sorafenib target Raf1, among other kinases [77]. Comparison of the response to sorafenib between HBV- and HCV-infected patients in the SHARP and Asian studies has not been published. The confounding possibility that HCV infection in itself confers a survival advantage is unlikely given that no differences in survival were noted in the placebo group of the SHARP study [73]. Despite these uncertainties, sorafenib is indicated for advanced HCC regardless of the etiology of underlying liver disease.
Sorafenib is now a standard of care in the first line treatment of advanced HCC. However, it should be noted that its efficacy has only been established in a highly selected patient population, >95 % of whom had Child-Pugh A cirrhosis [73, 74]. Although a pharmacokinetic analysis in the initial phase II study of sorafenib did not reveal any significant differences between Child-Pugh A and B patients, Child-Pugh B patients more frequently developed signs of hepatic decompensation on therapy [79]. A phase I pharmacokinetic study of sorafenib in a cohort of patients with various neoplasms reported a higher propensity for hyperbilirubinemia among those with hepatic dysfunction, but suggested that it was still possible to treat these patients, albeit at lower doses [80]. For example, patients with a serum bilirubin 1.5–3 times the upper limit of normal are recommended to receive half the daily dose of sorafenib, i.e., 400 mg daily. The study did not identify a safe dose for bilirubin above 3 times the upper limit of normal. The tolerance and safety of sorafenib in those with more severe hepatic dysfunction has not been formally tested. Retrospective studies of poor risk patients including those with Child-Pugh B/C cirrhosis and Child-Pugh A cirrhosis with portal vein thrombosis have reported grade 3–4 toxicities in 20–34 % of patients, leading to treatment discontinuation or dose reduction in up to 78 % of cases [81, 82]. Furthermore, those with more advanced cirrhosis experience shorter survival times on sorafenib than their counterparts with preserved liver function [79, 82].
In an attempt to improve upon the results seen with sorafenib alone, investigators have looked at combinations regimens with chemotherapy and other biologics. Doxorubicin, the historical chemotherapy backbone used in advanced HCC in the presorafenib era, was evaluated alone or with sorafenib in a randomized phase II study [83]. Both time-to-progression and progression-free survival were increased by approximately 4 months, and median overall survival doubled (13.7 versus 6.5 months, p = 0.006) with the sorafenib plus doxorubicin combination. Major grade 3–4 toxicities of patients on the combination arm were constitutional symptoms, pain, cutaneous reactions including hand-foot syndrome, gastrointestinal symptoms, and neutropenia without fever. A significantly higher percentage of patients on the combination arm experienced some degree of left ventricular systolic dysfunction while on study therapy (19 versus 2 %). One patient with no cardiac history developed grade 3–4 left ventricular dysfunction on sorafenib and doxorubicin. No grade 3–4 cardiac events occurred on the doxorubicin monotherapy arm. The study investigators acknowledged that the frequency of cardiac events occurring at a median cumulative doxorubicin dose of 165 mg/m2 warranted further investigation, and recommended that close cardiac monitoring be undertaken in patients receiving both drugs.
The synergistic effect of doxorubicin and sorafenib appears to occur through modulation of the proapoptotic pathway and evasion of the multi-drug resistance (MDR) pathway Fig. 2. Anthracycline-induced cytotoxicity is mediated by the proapoptotic kinase ASK1 [84]. Basic fibroblast growth factor (bFGF) activity leads to the expression of Raf-1 which binds to and neutralizes ASK1, thereby protecting the cell from apoptosis. Inhibition of Raf-1 by sorafenib would therefore be expected to restore chemosensitivity to doxorubicin. Another consequence of Raf activation is expression of the MDR-1 pump [85]. In vitro studies have shown that sorafenib appears to decrease ATP-binding cassette (ABC)/MDR-protein gene expression, resensitizing cells to gemcitabine as well as doxorubicin [86]. A randomized phase III study of sorafenib ± doxorubicin is currently underway (www.clinicaltrials.gov, NCT01015833). Sorafenib continues to be evaluated in combination with other chemotherapy drugs including gemcitabine, fluoropyrimidines, and platinums (www.clinicaltrials.gov, NCT00808145, NCT00844688, NCT00703365, NCT00941967, NCT01214343, NCT00933816, NCT01131689, NCT00752063, NCT01032850).
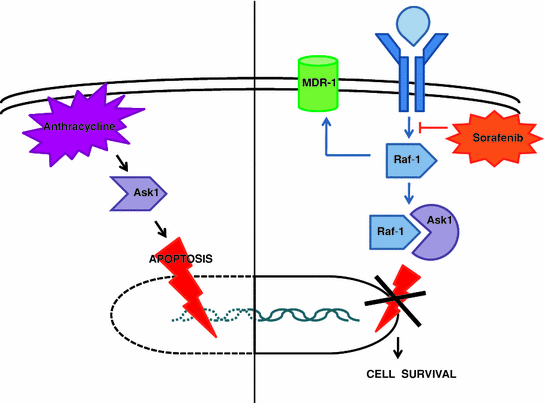
Fig. 2
Anthracyclines, Ask1 mediated apoptosis & sorafenib. MDR-1: Multi-durg resistance-1 pump; bFGF: Basic fibroblast growth factor
A phase I study looked at sorafenib and erlotinib in 17 patients with various solid tumors, including a single case of HCC [87]. There were 3 partial responses and 9 had stable disease, including the patient with HCC. All patients experienced mild to moderate fatigue, and 71–88 % experienced diarrhea, hypophosphatemia, and acneiform rash. A phase III trial is now investigating sorafenib in combination with erlotinib versus sorafenib alone in patients with advanced HCC only (www.clinicaltrials.gov, NCT00901901). A phase I-II study of dual antiangiogenic therapy with sorafenib and bevacizumab was suspended for undefined reasons (www.clinicaltrials.gov, NCT00867321). Sorafenib is also being paired with IGF-1R inhibitor IMC-A12 (www.clinicaltrials.gov, NCT00906373) and the angiopoietin inhibitor AMG386 (www.clinicaltrials.gov, NCT00872014) in phase II trials.
Sunitinib is another oral multitargeted kinase inhibitor similar to sorafenib. An initial phase II study of sunitinib monotherapy reported partial responses in 2.9 % of patients, stable disease in 50 %, and median progression-free survival and overall survival times of 3.9 and 9.8 months, respectively [88]. Grade 3–4 toxicities occurred in 3–18 % of patients and included neutropenia, thrombocytopenia, elevation of transaminases, and fatigue. Two deaths also occurred and were attributed to rapid disease progression and hepatic failure. Decreases in serum inflammatory biomarkers such as IL-6 and c-kit, as well as in vascular permeability as seen on dynamic contrast-enhanced (DCE)-MRI were significantly associated with disease control and survival. A second phase II study evaluated sunitinib at a higher dose, but was prematurely terminated after producing an objective response rate of 2.7 % [89]. There was a higher incidence of grade 3–4 toxicities in this study with myelosuppression, asthenia, and hand-foot syndrome occurring in 10.8–37.8 % of patients. There was also a 10.8 % toxic death rate. The failure of sunitinib to meet its primary endpoint of noninferiority, as well as the higher incidence of serious adverse events observed on the sunitinib arm led to the early termination of a randomized phase III trial against sorafenib (www.clinicaltrials.gov, NCT00699374). Sunitinib continues to be evaluated in a phase II trial with capecitabine (www.clinicaltrials.gov, NCT00787787).
Brivanib, an oral dual inhibitor of VEGF and FGF signaling, has shown activity in both untreated patients, and those who have failed antiangiogenic therapies such as sorafenib and thalidomide [90]. Disease control rates were similar in treatment-naïve and sorafenib pretreated patients at 47 and 53 %, as were time to progression at 2.8 and 2.0 months, respectively. The median overall survival for treatment-naïve patients was 10 months. Brivanib appears to be tolerable even after prior antiangiogenic therapy [91], and those who achieve disease control by modified RECIST criteria experience better survival outcomes [92].
Linifanib is an oral inhibitor of VEGF and PDGFR receptor tyrosine kinases. A phase II trial was conducted in 44 patients with Child-Pugh A and B cirrhosis. The primary endpoint, 16 week progression-free-survival rate, was 42.1 %. There was an overall response rate of 8.7 % in the Child-Pugh A cohort, both the median time-to-progression and progression-free survival were 112 days, and the median overall survival was 295 days on preliminary analysis [93]. No patients with Child-Pugh B cirrhosis responded to linifanib. Updated results presented for the total cohort of 44 patients revealed an overall 16 week progression-free survival rate of 31.8 %, overall response rate of 6.8 %, median radiographic time-to-progression of 5.4 months, and overall survival of 9.7 months [94]. As in the preliminary report, the presence of Child-Pugh B cirrhosis was associated with a lesser degree of benefit from linifanib. The most frequent treatment-related grade 3–4 adverse events were hypertension and fatigue, and 68 % of patents had interruptions in treatment due to reversible hypertension and proteinuria. One toxic death due to intracranial hemorrhage occurred in a patient with Child-Pugh B cirrhosis. Linifanib is now being evaluated against sorafenib in a phase III trial (www.clinicaltrials.gov, NCT0100959).
Vatalanib is an oral small-molecule TKI of the VEGFR which, at higher concentrations, also inhibits PDGFR, c-Kit and c-Fms [95]. Preclinical studies in mice showed that vatalanib inhibits tumor growth by decreasing microvessel formation [89], cell proliferation, and by promoting apoptosis [96]. A phase I clinical trial showed vatalanib to be tolerable in patients with hepatocellular and mild degrees of hepatic dysfunction. No objective responses were recorded although 50 % of evaluable patients had disease stabilization [97].
Cediranib, or AZD2171, inhibits VEGFR-2 and has a similar mechanism of action as vatalanib [98]. An interim safety analysis of phase I trial of AZD2171 in HCC reported an 84 % incidence of grade 3 toxicities consisting of fatigue, hypertension, and anorexia, with treatment discontinuation due to patient refusal in 29 % of cases [99].
1.3 Intracellular Signaling Cascades
1.3.1 The Ras/Raf/Mek/Erk Pathway
This ubiquitous signaling cascade drives key processes for normal cell growth, proliferation, and survival [100], and is also involved in tumorigenesis and metastases [101]. Ras and Raf are the main regulators of this pathway, and also interact with the mTOR and phospholipase Cx signal transduction pathways [9, 53, 102]. The overexpression and constitutive activation of various components of this pathway has been implicated in hepatocarcinogenesis, tumor cell survival, and disease progression [9, 100, 103], with a concurrent downregulation of inhibitory proteins [104, 105]. Precipitants of Ras/Raf/Mek/Erk dysregulation include infection with viral hepatitis [77, 106, 107]. As previously mentioned, HCV induces the upregulation of Raf1 which may explain the improved outcomes seen among these patients in response to sorafenib [78]. Mutations of pathway components occur less frequently at a rate of approximately 3–4 % for Ras and Raf [13]. Occupational exposures such as vinyl chloride, associated with autoclave cleaning, have been associated with a higher prevalence of K-ras-2 mutations [108].
Carboxy-terminal farnesylation is the final processing step necessary to generate functional Ras [109, 110]). Direct farnesyltransferase inhibition has shown chemopreventive activity in vitro [111], but has not yet been tested clinically. HMG CoA reductase is a component of the mevalonate biosynthesis pathway which also produces farnesyl [112]. Diabetic patients treated with statins, which are HMG CoA reductase inhibitors, have been observed to have a significantly reduced risk of developing HCC compared to matched controls [113]. Furthermore, the addition of pravastatin to standard therapy for advanced HCC doubled the overall survival time (18 versus 9 months, p=0.006) in a single institution randomized controlled trial [114]. These intriguing findings have led to an ongoing randomized phase III study of sorafenib ± pravastatin (www.clinicaltrials.gov, NCT01075555). A phase IB trial of the direct farnesyltransferase inhibitor SCH 66336 ± gemcitabine in the neoadjuvant setting for patients with resectable HCC has been completed with results pending (www.clinicaltrials.gov, NCT00020774).
In vitro studies of the Mek 1–2 inhibitor, AZD6244, have been shown to decrease HCC tumor growth in a dose-dependent fashion, through inhibition of Erk 1–2 and upregulation of apoptosis-mediating caspases [115]. The addition of doxorubicin to AZD6244 appeared to have a synergistic effect through upregulation of the p130 RB tumor suppressor gene [115]. AZD6244 is now being assessed clinically in HCC as monotherapy in a phase II study (www.clinicaltrials.gov, NCT00550719), and with sorafenib in a phase I/II study (www.clinicaltrials.gov, NCT01029418). A phase II trial evaluating sorafenib with another Mek inhibitor, BAY86-9766 in patients with HCC is expected to open soon (www.clinicaltrials.gov, NCT01204177).
1.3.2 The PI3K/Akt/mTOR Pathway
The mammalian target of rapamycin (mTOR) sits downstream of the PI3K/Akt pathway which is regulated by the phosphatase and tensin homolog deleted on chromosome 10 (PTEN) tumor suppressor gene [116]. By integrating signals from an array of mitogens including EGF, IGF, VEGF, PDGF, and HIF-1α [9, 53, 117, 118], mTOR activation exerts proangiogenic [119], proproliferative [120], and antiapoptotic effects ([53, 121]). Up to 50 % of human HCC tumor cells have been shown to exhibit deviant mTOR activity in conjunction with upregulated EGFR and IGF-1R signaling, and PTEN downregulation [118]. Increased markers of mTOR activity also appear to have prognostic relevance; tumors tend to be larger and more poorly differentiated, time to recurrence is significantly decreased [118] as is overall survival among R0-resected patients [122]. For all of these reasons, mTOR is a potential target to be evaluated in HCC .
In vitro and in vivo models of mTOR inhibition in HCC have shown that tumor regression occurs through disruption of cell cycling and proliferation, but not by induction of apoptosis [118]. Retrospective observations of improved survival among patients who received sirolimus immunosuppression following liver transplantation for HCC [123–126] have also provided the basis for several clinical trials of mTOR inhibition in this disease. A small phase II study of sirolimus in 21 and 9 patients with advanced HCC and cholangiocarcinoma reported a single partial response, 5 with disease stabilization, and a median survival of 6.5 months in the HCC group [127]. Everolimus monotherapy is being evaluated in two first line phase I-II studies (www.clinicaltrials.gov, NCT00390195, NCT00516165), and as second line therapy post-sorafenib failure in the phase III randomized, placebo-controlled EVOLVE-1 study (www.clinicaltrials.gov, NCT01035229). The benefit of adding everolimus to doxorubicin-based TACE in patients with liver-confined HCC is also being assessed in a phase I/II study (www.clinicaltrials.gov, NCT01009801). The safety and efficacy of temsirolimus in patients with advanced HCC and Child-Pugh B cirrhosis is the subject of a phase II study (www.clinicaltrials.gov, NCT01079767). AZD8055 is another oral mTOR inhibitor currently being tested in two phase I and II trials (www.clinicaltrials.gov, NCT00999882, NCT00973076). A phase III trial is evaluating post-liver transplant outcomes with sirolimus vs tacrolimus (www.clinicaltrials.gov, NCT00554125).
Given the known cross-communication between the mTOR and angiogenic pathways, combined inhibition is an appealing strategy. Phase I and II trials of everolimus with sorafenib (www.clinicaltrials.gov, NCT00828594, NCT01005199) and bevacizumab (www.clinicaltrials.gov, NCT00775073; [117]) in advanced HCC are ongoing. A phase I trial of temsirolimus and sorafenib is currently recruiting, and the safety of this combination in patients with liver dysfunction will be evaluated in an as yet unopened phase I trial (www.clinicaltrial.gov, NCT01008917, NCT01013519). Temsirolimus is also being studied in combination with bevacizumab in a phase II trial including various solid tumors, including HCC (www.clinicaltrials.gov, NCT01010126). Furthermore, sirolimus is also being combined with bevacizumab in a phase I study enrolling patients with unresectable HCC (www.clinicaltrials.gov, NCT00467194).
1.4 Evaluating Responses to Targeted Therapies
Unlike the vast majority of chemotherapy drugs which are cytotoxic, many of these targeted agents are cytostatic, given their inhibitory effects on cell cycling, proliferation, and tumor angiogenesis. Consideration of these concepts has led to the recognition that responses to these very different types of treatments cannot be assessed in the same manner. Conventional Response Evaluation Criteria in Solid Tumors (RECIST) guidelines evaluate changes in two-dimensional tumor measurements to classify the extent and direction of response from baseline as a result of cytotoxic chemotherapy [128]. However, this paradigm may not accurately characterize responses to cytostatic biologic therapies where decreases in tumor viability, an arguably more important reflection of treatment activity, are not necessarily associated with tumor shrinkage or disappearance [70, 72, 129]. New response assessment tools have been developed in an attempt to more adequately capture responses to targeted agents and predict survival outcomes. The concept of tumor viability, reflected by tissue density due to vascular contrast enhancement, is a key element of these new guidelines. Measurements of changes in tumor density have contributed to the success of the Choi criteria in patients with gastrointestinal stromal tumors treated with imatinib, which outperformed RECIST guidelines in response detection and correlation with survival outcomes [130]. As previously mentioned, in the initial phase II trial of sorafenib, concurrent increases in both central tumor necrosis and tumor size due to inflammation and edema were observed in responding patients [70]. The ratio of tumor necrosis relative to tumor volume was shown to increase significantly from baseline in a subset of patients who responded to sorafenib compared to those who did not [72]. The application of this model as a response assessment tool requires validation on a larger prospective trial. Currently, the modified RECIST (mRECIST) guidelines use decreases in intratumoral enhancement to define a response [131–133]. An important caveat is that changes in intratumoral enhancement may reflect changes in tumor vasculature simply as a result of exposure to antiangiogenic agents, and not be indicative of antitumoral activity [133]. For example, the lack of an overall survival benefit despite an apparent radiographic response to antiangiogenic agents such as bevacizumab and cediranib in patients with malignant gliomas has led investigators to update and modify response criteria and study endpoints [134, 135]. Prospective analyses and histologic confirmation of tumor cell death will therefore be an important means to corroborate radiologic findings.
Functional imaging modalities may eventually help to further evaluate response assessment in HCC. For example, dynamic contrast-enhanced MRI (DCE-MRI) has shown real-time decreases in arterial enhancement which correlate with declines in circulating proangiogenic factors in HCC patients treated with bevacizumab [64]. Similarly, in the phase II sunitinib study, decreases in vascular permeability on DCE-MRI as well as serum proinflammatory and proangiogenic factors, appeared to be associated with better outcomes [88]. However, in a study of 6 patients receiving doxorubicin and sorafenib, changes in tumor vascularity and permeability on DCE-MRI using a computer algorithm did not correlate significantly with responses [136]. Larger studies need to be conducted in order to better evaluate the validity of DCE-MRI as a response evaluation modality. Newer MRI imaging techniques such as blood oxygen level dependent (BOLD) [137], diffusion weighting [138], and image subtraction [139] have shown promise in assessing tumor necrosis in response to ablative therapies, and could potentially be applicable for systemic antiangiogenic therapies.
Related posts:
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


