The concept of using monoclonal antibodies for delivering drugs to cancer cells has been explored for decades, with early work surrounding nonspecific targets and drugs with low potencies. These studies underscored the importance of critical parameters, such as antigen and tumor target selection, linker stability, drug potency, pharmacokinetics, and conjugation methodology, in developing effective antibody drug conjugates with acceptable safety profiles. Brentuximab vedotin represents the culmination of much research and development activities in which many of these parameters were addressed. This article provides an overview of many studies that led to the development of this highly active antibody drug conjugate.
Key points
- •
The concept of using monoclonal antibodies for delivering drugs to cancer cells has been explored for decades, with early work surrounding nonspecific targets and drugs with low potencies.
- •
These studies underscored the importance of critical parameters, such as antigen and tumor target selection, linker stability, drug potency, pharmacokinetics, and conjugation methodology, in developing effective antibody drug conjugates with acceptable safety profiles.
- •
Brentuximab vedotin represents the culmination of much research and development activities in which many of these parameters were addressed.
- •
The drug received accelerated approval for the treatment of relapsed Hodgkin lymphoma and systemic anaplastic large cell lymphoma.
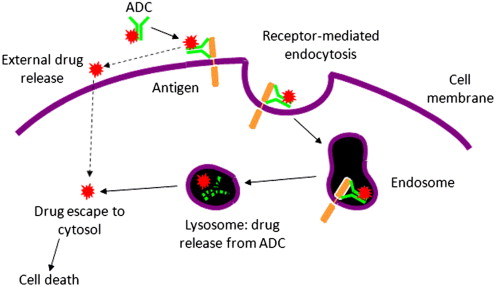
Antibody–drug conjugates (ADCs) for cancer therapy are designed for the specific delivery of cytotoxic agents to tumor tissues, ideally allowing for selective ablation while sparing normal cells from chemotherapeutic damage. The success of an ADC is driven by several factors, not least of which is the choice of appropriate antigen and tumor target, the monoclonal antibody (mAb) used for delivery, the cytotoxic agent attached to the mAb, and the linker and conjugation technology used to join the 2 components ( Fig. 1 ). The highly specific binding interaction between the mAb and the antigen of interest, and the expression profile of the antigen, are primary drivers of preferential delivery to tumor cells. Drug potency and the mode and multiplicity of attachment to the mAb are also strong determinants in achieving specific cell kill. All of these factors were addressed in the development of brentuximab vedotin (ADCETRIS), an ADC composed of an anti-CD30 mAb (cAC10) linked to monomethyl auristatin E (MMAE) through a protease-cleavable linker. Brentuximab vedotin received accelerated approval by the U.S. Food and Drug Administration for the treatment of relapsed Hodgkin lymphoma and systemic anaplastic large cell lymphoma (ALCL). This article details some of the technological advancements and preclinical and clinical findings that led to the generation of this drug.
The CD30 target antigen
Cancer cells often overexpress cell surface antigens that distinguish them from normal tissues and allow for the use of targeting reagents for therapeutic intervention. Many of these antigens have been exploited using mAbs designed to recognize them, bind, and elicit selective cell kill because of effector function and signaling activities. To date, 9 mAbs directed against target antigens on leukemias, lymphomas, tumor-infiltrating lymphocytes, and solid tumor malignancies have been clinically approved ( Table 1 ). The clinical impact of these approved agents has spawned great interest in exploiting other antigens that are selectively expressed within tumors. CD30 is an example of a highly desirable antigen for targeted therapy, because of minimal normal tissue expression coupled with high expression on some human cancers. For example, this antigen is highly expressed on the Reed-Sternberg cells of Hodgkin lymphoma and on systemic ALCL tumor cells. CD30 is also observed on cutaneous T-cell lymphoma and other select lymphoid tumors and some malignancies, including germ cell cancers. Its normal physiologic expression is otherwise thought to be limited to activated T and B cells. CD30 is a tumor necrosis factor receptor (TNFR) superfamily member, which stimulates apoptosis via TNFR-associated factor 2 degradation. Because of its expression profile and its role in the regulation of cell survival, considerable attention has been given to CD30 as a target both for anti-CD30 mAbs and for ADCs. Several studies have been reported for anti-CD30–based therapies, including work with an unconjugated mAb, and anti-CD30–toxin conjugates. The unconjugated anti-CD30 mAb, cAC10, had promising preclinical activity profiles based on its ability to retard tumor outgrowth through antibody-dependent cellular phagocytosis (ADCP) and signaling activities, However, unconjugated cAC10 failed to show convincing clinical efficacy. Much more potent anti-CD30–based reagents were generated with a mAb-ricin A chain conjugate and a mAb-saporin conjugate. Although these conjugates had very promising in vitro and in vivo activities, they, like many other conjugates such as these, failed to generate convincing clinical efficacy, mostly because of immunogenicity and issues with respect to targeted delivery. Taken together, these studies provided strong preclinical data supporting CD30 as a target for an antibody-based therapy, but indicated that improvements were needed to effectively exploit the presence of this antigen on cancer cells.
| Target | Antibody | Therapeutic Indication | First U.S. Approval |
|---|---|---|---|
| CD20 | Rituximab | Non-Hodgkin’s lymphoma | 1997 |
| CD20 | Ofatumumab | Chronic lymphocytic leukemia | 2009 |
| Her2 | Trastuzumab | Breast cancer | 1998 |
| Her2 | Pertuzumab | Breast cancer | 2012 |
| CD52 | Alemtuzumab | Chronic lymphocytic leukemia | 2001 |
| EGFR | Cetuximab | Colon cancer | 2004 |
| EGFR | Panitumumab | Colon cancer | 2006 |
| VEGF | Bevacizumab | Colon cancer | 2004 |
| CTLA-4 | Ipilimumab | Melanoma | 2011 |
The CD30 target antigen
Cancer cells often overexpress cell surface antigens that distinguish them from normal tissues and allow for the use of targeting reagents for therapeutic intervention. Many of these antigens have been exploited using mAbs designed to recognize them, bind, and elicit selective cell kill because of effector function and signaling activities. To date, 9 mAbs directed against target antigens on leukemias, lymphomas, tumor-infiltrating lymphocytes, and solid tumor malignancies have been clinically approved ( Table 1 ). The clinical impact of these approved agents has spawned great interest in exploiting other antigens that are selectively expressed within tumors. CD30 is an example of a highly desirable antigen for targeted therapy, because of minimal normal tissue expression coupled with high expression on some human cancers. For example, this antigen is highly expressed on the Reed-Sternberg cells of Hodgkin lymphoma and on systemic ALCL tumor cells. CD30 is also observed on cutaneous T-cell lymphoma and other select lymphoid tumors and some malignancies, including germ cell cancers. Its normal physiologic expression is otherwise thought to be limited to activated T and B cells. CD30 is a tumor necrosis factor receptor (TNFR) superfamily member, which stimulates apoptosis via TNFR-associated factor 2 degradation. Because of its expression profile and its role in the regulation of cell survival, considerable attention has been given to CD30 as a target both for anti-CD30 mAbs and for ADCs. Several studies have been reported for anti-CD30–based therapies, including work with an unconjugated mAb, and anti-CD30–toxin conjugates. The unconjugated anti-CD30 mAb, cAC10, had promising preclinical activity profiles based on its ability to retard tumor outgrowth through antibody-dependent cellular phagocytosis (ADCP) and signaling activities, However, unconjugated cAC10 failed to show convincing clinical efficacy. Much more potent anti-CD30–based reagents were generated with a mAb-ricin A chain conjugate and a mAb-saporin conjugate. Although these conjugates had very promising in vitro and in vivo activities, they, like many other conjugates such as these, failed to generate convincing clinical efficacy, mostly because of immunogenicity and issues with respect to targeted delivery. Taken together, these studies provided strong preclinical data supporting CD30 as a target for an antibody-based therapy, but indicated that improvements were needed to effectively exploit the presence of this antigen on cancer cells.
| Target | Antibody | Therapeutic Indication | First U.S. Approval |
|---|---|---|---|
| CD20 | Rituximab | Non-Hodgkin’s lymphoma | 1997 |
| CD20 | Ofatumumab | Chronic lymphocytic leukemia | 2009 |
| Her2 | Trastuzumab | Breast cancer | 1998 |
| Her2 | Pertuzumab | Breast cancer | 2012 |
| CD52 | Alemtuzumab | Chronic lymphocytic leukemia | 2001 |
| EGFR | Cetuximab | Colon cancer | 2004 |
| EGFR | Panitumumab | Colon cancer | 2006 |
| VEGF | Bevacizumab | Colon cancer | 2004 |
| CTLA-4 | Ipilimumab | Melanoma | 2011 |
Development of the drug-linker component for a targeted anti-CD30 therapeutic
The concept of using mAbs for delivering drugs to tumor cells was proposed long before tumor-selective mAbs were available. With the advent of newer technologies for the generation of selective, nonimmunogenic mAbs, the field began to be extensively explored using approved anticancer drugs, such as doxorubicin and vinca alkaloids. Clinical trials with drugs such as BR96-doxorubicin highlighted some underlying weaknesses of this approach, which included toxicity from normal tissue antigen expression; low potency of the drug component, requiring high doses to be administered; and the instability of the linker used between the drug and the mAb, resulting in much of the drug being lost within the first few days of administration. To overcome some of these issues, attention turned to the use of highly potent cytotoxins targeted with mAbs that displayed higher degrees of tumor specificity. Gemtuzumab ozogamicin (Mylotarg) was one such agent, which incorporated a high-potency drug component (calicheamicin), conjugated to an antibody (anti-CD33) that displayed significant specificity for antigens expressed on acute myelogenous leukemia. Although the agent was found to be clinically active, the utility of the drug may have been hampered by the instability of the linker, the susceptibility of the drug to multidrug resistance, and the presence of large amounts of unconjugated antibody in the drug product. Although gemtuzumab ozogamicin was being developed and investigated, continued efforts with highly potent immunotoxins continued, but the efforts were thwarted because of immunogenicity and toxicity. Thus, a newer-generation ADC technology required the development of potent drugs that were nonimmunogenic, and could be conjugated to highly selective antitumor antibodies through linker technologies that were more stable than those used previously.
In generating an appropriate drug for ADCs, the authors focused attention on totally synthetic drugs that could be tailor-made for drug delivery. This research led to the development of MMAE ( Fig. 2 ), a synthetic analog of dolastatin 10, which was one of several toxic natural products isolated from the Indian Ocean sea hare, Dolabella auricularia . MMAE exerts its anticancer activity through the inhibition of tubulin polymerization. Some of the features that make MMAE appropriate as the cytotoxic drug component of an ADC include a high potency against a broad array of tumor cell lines; its high water solubility, which helps circumvent ADC aggregation; its stability under physiologic conditions; and, importantly, an intentionally built-in site for stable linker attachment to enable mAb conjugation.
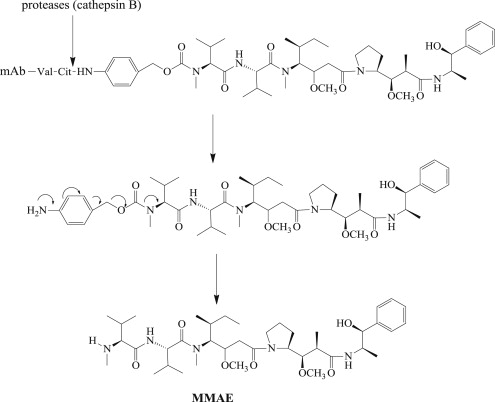
A great deal of research has surrounded how MMAE could be effectively conjugated to mAbs against cancer cell–surface antigens, with specific attention placed on the cAC10 mAb that recognizes the CD30 antigen on various lymphomas. Historically, there are many ways to attach drugs to mAbs, but most involve the reaction of cysteine side chains with thiol-reactive moieties or lysine side chain amine reactions with active esters. The presence of 4 reducible disulfide bonds within the immunoglobulin G (IgG) structure that covalently link the heavy and light chains together is an attractive feature that allows for selective attachment of drug to up to 8 distinct sites using a thiol-reactive handle. Up to 100 lysines are present in typical IgG1 mAbs, and conjugation to these sites through amine-reactive linkers results in a great deal more heterogeneity than that obtained using cysteines generated through disulfide reduction ( Fig. 3 ).
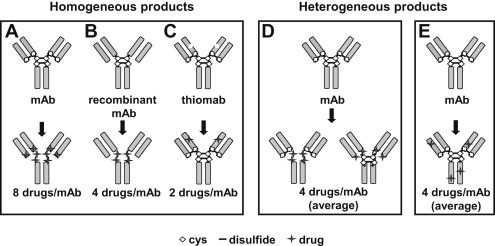
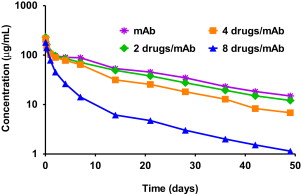
The number of drugs conjugated to reduced antibody disulfides can be optimized using partial reduction techniques. Although in vitro cytotoxicity potency increases with increased multiplicity of drug conjugation, in vivo potency is actually diminished. This paradoxic observation can be explained by ADC pharmacokinetics, in which it was found that longer circulation half-lives were obtained with drug loadings of 2 or 4 per mAb compared with fully loaded ADCs containing 8 drugs per mAb ( Fig. 4 ). Controlling drug substitution to levels that preserve the pharmacokinetic parameters of the parent mAb has generally been found to provide longer tumor exposure, better activity, and reduced toxicity.