The identification of the intrinsic molecular subtypes of breast cancer has enhanced our understanding of tumor biology, informing therapeutic targets, and clinical trial design. This article reviews the intrinsic classification system and the clinically defined subtypes of breast cancer. We review the molecular drivers of each subtype and discuss implications for prognosis, clinical management, and future directions.
Key points
- •
The intrinsic subtypes of breast cancer have enhanced understanding of the molecular drivers underlying subgroups of tumors.
- •
Endocrine receptor–positive breast cancer may develop resistance to antiestrogen therapy, and therapies to overcome this resistance are being developed.
- •
The management of HER2 (human epidermal growth factor receptor 2)-positive breast cancer has been revolutionized by the development of HER2 targeting drugs.
- •
In triple-negative breast cancer, there are no currently validated targeted therapies; however, several novel approaches are being investigated.
Introduction
Breast cancer represents a complex and heterogenous group of diseases with distinct morphologic and molecular features. Most patients present with early-stage disease, and a proportion of these are cured with local therapy alone. Accurate identification of patients who are at risk for recurrent or metastatic disease, and the selection of appropriate systemic interventions for these patients, has helped to improve survival.
The risk of recurrence has traditionally been estimated by amalgamating clinicopathologic features, including tumor size, histologic grade, lymph node involvement, estrogen receptor (ER), progesterone receptor (PR), and human epidermal growth factor receptor 2 (HER2) receptor status. Patients stratified into a high-risk category are often advised to undergo systemic therapy; however, this approach overtreats some patients, exposing them to the potential for significant toxicity without corresponding benefit. In contrast, some tumors that have low-risk features may subsequently recur. It is clear that these clinicopathologic criteria incompletely represent underlying tumor biology. Significant effort has been made over the last decade to investigate the molecular drivers of breast tumors. Through better understanding of breast cancer biology, the ability to prognosticate and predict treatment benefit in early-stage disease, and to overcome resistance to therapy and improve survival in metastatic breast cancer (MBC), is becoming a reality.
Introduction
Breast cancer represents a complex and heterogenous group of diseases with distinct morphologic and molecular features. Most patients present with early-stage disease, and a proportion of these are cured with local therapy alone. Accurate identification of patients who are at risk for recurrent or metastatic disease, and the selection of appropriate systemic interventions for these patients, has helped to improve survival.
The risk of recurrence has traditionally been estimated by amalgamating clinicopathologic features, including tumor size, histologic grade, lymph node involvement, estrogen receptor (ER), progesterone receptor (PR), and human epidermal growth factor receptor 2 (HER2) receptor status. Patients stratified into a high-risk category are often advised to undergo systemic therapy; however, this approach overtreats some patients, exposing them to the potential for significant toxicity without corresponding benefit. In contrast, some tumors that have low-risk features may subsequently recur. It is clear that these clinicopathologic criteria incompletely represent underlying tumor biology. Significant effort has been made over the last decade to investigate the molecular drivers of breast tumors. Through better understanding of breast cancer biology, the ability to prognosticate and predict treatment benefit in early-stage disease, and to overcome resistance to therapy and improve survival in metastatic breast cancer (MBC), is becoming a reality.
Intrinsic subtypes of breast cancer
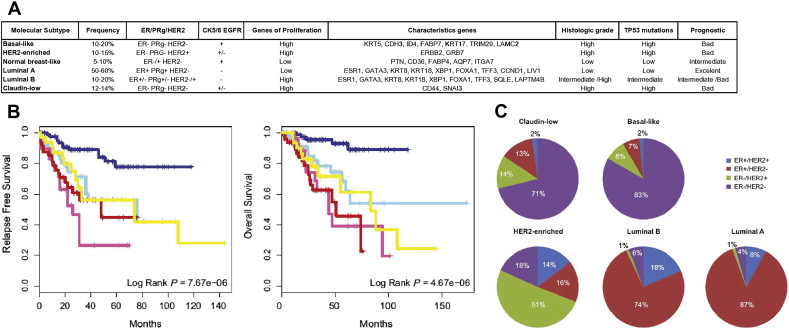
Perou and Sorlie and colleagues used gene expression profiling to group breast cancers into 5 distinct molecular classes or intrinsic subtypes, termed luminal A, luminal B, basal-like, HER2-enriched, and normal-like subtypes. A further subtype, claudin-low, was recently identified. This classification reflects clinical subdivisions based on ER and HER2 status but, independent of these variables, it identifies groups with differing survival and treatment responses ( Fig. 1 ).
Luminal A and Luminal B
Luminal A is the most common subgroup and accounts for 40% to 60% of breast cancers. These tumors are predominantly ER+/PR+ and HER2−, with low histologic grade and low expression of proliferative genes, including Ki67. They are associated with lower relapse rate and longer survival from the time of that relapse, compared with the other subtypes. Luminal B cancers, in common with luminal A, express high levels of luminal cytokeratins and are predominantly ER+, although they have lower expression of ER-related genes. They make up 15% of breast cancers and, in contrast with luminal A tumors, are more aggressive, with significantly poorer prognosis. In addition, the TP53 pathway is conserved in luminal A but frequently inactivated in the luminal B subtype. Luminal B tumors are histologically high grade and have increased expression of proliferation genes (eg, Ki67 and cyclin B1) with variable PR and HER2 status. The management of luminal A tumors is centered on antiendocrine strategies, and this subtype is less responsive to chemotherapy. Although luminal B tumors are also ER+, a significant number do not respond to antiestrogen therapy and instead show greater response to chemotherapy.
HER2 Enriched
The HER2-enriched subtype makes up 10% of breast cancers and is characterized by high expression of HER2 and related genes. This subtype is associated with increased coexpression of proliferative genes and has high genomic instability. Although chemosensitive, the HER2-enriched subtype was a poor prognosis category until the introduction of HER2-targeted drugs. There is incomplete concordance between histologic and intrinsic subtyping, with only 70% of HER2-enriched tumors by microarray showing protein overexpression. For tumors that are HER2 enriched but clinically HER2−, the role of anti-HER2 therapy is unclear. In addition, a significant number of clinically HER2+ tumors belong to the luminal B cluster, and these are associated with better prognosis than those that are HER2 enriched.
Basal-like
Basal-like breast cancers (BLBCs) express genes characteristic of normal breast myoepithelial cells, such as cytokeratins 5, 6, and 17. These tumors represent 10% to 25% of breast cancers, are highly proliferative, and have a high rate of p53 mutations. Basal-like tumors are associated with increased genetic complexity and are purported to be less stable compared with other breast subtypes. They do not express ER/PR or HER2, and overlap with the clinically defined triple-negative breast cancers (TNBCs). However, these terms are not synonymous. Not all tumors with a basal-like gene expression profile are triple negative; 15% to 45% express ER and HER2 or both. In contrast, only 85% of TNBCs have a basal-like molecular phenotype. BRCA1 breast cancers are associated with the basal-like subgroup. Although these tumors are chemoresponsive, they have shown a poor prognosis across several studies.
Claudin-low
Claudin-low tumors make up 7% to 14% of breast cancers and have low expression of genes involved in cell-cell adhesion, including claudin 3, 4, 7, and E-cadherin. These tumors have metaplastic and medullary differentiation, and they predominantly present as TNBC. However, there is incomplete concordance, with 15% of claudin-low breast cancers expressing ER and 15% overexpressing HER2. These tumors may also be associated with BRCA1 mutations. Claudin-low tumors do not show high expression of proliferation genes, but they also have a poor prognosis. These tumors are unique in being enriched for epithelial-to-mesenchymal transition markers, and have a significant immune cell infiltrate and cancer stem cell–like features. Claudin-low tumor response to chemotherapy is intermediate between that of basal-like and luminal subtypes.
Normal-like
The normal-like subset expresses genes characteristic of adipose tissue and represents 3% to 10% of breast cancers. These tumors are frequently ER+ and have low levels of proliferative genes and low tumor cellularity. The prognosis of this group is reported to be intermediate, with better survival than all but luminal A breast cancers. There is controversy as to the significance of this group, and it has been suggested that it represents technical artifact.
The identification of these molecular signatures has reshaped understanding of breast cancer and helps to inform the search for novel therapies. However, this classification is a work in progress, requiring refinement and standardization before it can be incorporated into clinical practice and decision making. Further intrinsic subtypes, such as interferon-rich and molecular apocrine, have been proposed, but the significance of these groups remains unknown.
Several tools have been developed in an attempt to use molecular profiling to prognosticate and predict benefit from therapy. The PAM50 assay classifies tumor samples into intrinsic subtypes and predicts risk of relapse. However, it lacks validation and is not ready for clinical decision making. Several multigene signatures have also been developed that can risk stratify and predict therapeutic response to varying degrees. These multigene signatures include MammaPrint, OncotypeDx, Theros/MGI, MapQuant Dx, Rotterdam/Veridex 76-gene, and the wound-response gene signatures. MammaPrint is a prognostic test that has been granted US Food and Drug Administration approval for assessment of breast cancer recurrence risk in tumors that are lymph node negative, less than 5 cm, and either ER positive or ER negative. OncotypeDx applies to ER-positive, primarily node-negative breast cancer, and can predict risk of recurrence and likelihood of benefit from chemotherapy. It has also shown similar predictive power in retrospective analysis of lymph node–positive patients. Both tests are currently in clinical use; however, the Evaluation of Genomic Applications in Practice and Prevention (EGAPP) working group assessment found that further evaluation of these technologies is required. Prospective validation is ongoing with the MINDACT (MammaPrint), TAILORx (OncotypeDx), and RxPONDER (OncotypeDx, in patients with 1–3 positive lymph nodes) studies.
Therefore, despite the progress and better understanding of the drivers of this disease, from a clinical management perspective, breast cancer remains divided into 3 therapeutic categories:
- 1.
ER+ disease, which is targeted with antiendocrine strategies
- 2.
HER2+ disease, which is treated with HER2-targeted agents
- 3.
Triple-negative breast cancer, which lacks validated targeted therapy options and is treated with traditional cytotoxic therapy.
This article discusses how translational research has led to the development of targeted therapies based on the drivers of specific subgroups of breast cancer.
ER-positive breast cancer
Endocrine Therapy
Targeting the ER is well established in the treatment of early and advanced ER+ breast cancer. This approach was pioneered in the 19th Century when Beatson noted tumor responses in women with advanced breast cancer treated with oophorectomy. With the identification of the ER, it was subsequently possible to select patients likely to benefit from antiendocrine therapy. Tamoxifen, a selective ER modulator, was approved in the late 1970s for the treatment of postmenopausal women with MBC. Later, its role in preventing disease recurrence was established, and tamoxifen was incorporated into adjuvant therapy, for all women with ER+ breast cancer. In the mid-1990s, anastrozole was the first aromatase inhibitor (AI) developed for postmenopausal women with ER+ MBC. Shortly afterward, it too was approved for adjuvant use. Other AIs, letrozole and exemestane, followed and are applied in both the metastatic and adjuvant settings. The AIs have shown increased efficacy compared with tamoxifen, but their benefit is limited to postmenopausal women based on their mechanism of action, which causes a paradoxic estrogen surge in women with functioning ovaries. Fulvestrant, a selective ER downregulator, was subsequently developed for postmenopausal women with progression of ER+ breast cancer following antiestrogen therapy. In premenopausal women, tamoxifen remains the first-line antiendocrine strategy in both the adjuvant and metastatic setting. The role of ovarian suppression has been explored, and, in premenopausal women with MBC, the combination of tamoxifen with ovarian suppression improves survival. The role of ovarian suppression in the adjuvant setting is less clear, and the results of the Suppression of Ovarian Function Plus Either Tamoxifen or Exemestane Compared With Tamoxifen Alone in Treating Premenopausal Women With Hormone-Responsive Breast Cancer (SOFT) study are awaited.
Despite these effective antiendocrine strategies, several patients develop treatment resistance—in adjuvant patients with the development of metastatic disease or, in the context of MBC, progression of disease in the face of ongoing therapy. This may be caused by de novo resistance (ie, having never responded to antiestrogen therapy) or acquired resistance shown by refractory disease following an initial period of response. Extensive efforts have been made to better elucidate mechanisms of resistance to identify potential therapeutic targets that may overcome this resistance.
Signaling downstream of ER involves multiple crosstalking and potentially compensatory pathways, including mitogen-activated protein kinase (MAPK), phosphatidylinositide 3-kinase (PI3K)/AKT/mammalian target of rapamycin (mTOR), epidermal growth factor receptor (EGFR), insulinlike growth factor receptor (IGF-IR), fibroblast growth factor (FGF) receptor, and Src ( Fig. 2 ). Inhibition of the ER may cause upregulation of these alternative pathways, driving resistance to therapy.
mTOR
Increased activity of PI3K/AKT is associated with endocrine resistance in breast cancer cells. There has been significant interest in targeting mTOR, a downstream mediator of this pathway, using inhibitors developed for transplant immunotherapy and kidney cancer. Inhibition of mTOR restored sensitivity to endocrine therapy in breast cancer cells. The randomized phase III study BOLERO-2 examined the combination of everolimus (an mTOR inhibitor) and exemestane versus exemestane alone in women with progressive metastatic disease following an AI. Everolimus significantly improved progression-free survival (PFS) from 4.1 to 10.6 months (hazard ratio, 0.36; 95% confidence interval [CI], 0.27 to 0.47; P <.001). This improvement came with additional toxicity, notably an increase in greater than or equal to grade 3 stomatitis, anemia, dyspnea, hyperglycemia, and pneumonitis. TAMRAD, a phase II study of tamoxifen with or without everolimus, also included women who had resistance to an AI. This trial met its primary end point of improved clinical benefit rate (6-month clinical benefit rate, 61% vs 42%). In addition, there was improved time to progression and overall survival (OS) with the combination supporting the benefit of mTOR inhibition seen in BOLERO-2. A phase I/II study of the combination of an alternative mTOR inhibitor, sirolimus, and tamoxifen similarly improved PFS in women who were previously treated with antiestrogen therapy and in the first-line setting.
In contrast, the HORIZON study, which combined the mTOR inhibitor, temsirolimus, with letrozole as first-line therapy for MBC, was terminated for futility in PFS. Patient population differences may account for these contrasting results, because women in BOLERO-2 and TAMRAD had endocrine-resistant disease, whereas the HORIZON study was conducted in the first-line setting. It is also possible that inadequate temsirolimus dosing may have contributed to the disappointing outcome because lower-than-anticipated adverse events were observed. Significant heterogeneity across ER+ breast cancers likely influences drivers of resistance and response to any given therapy. A more specific biomarker than ER status may be required to select patients who will respond to mTOR inhibition.
PIK3CA
There is also interest in targeting PI3K, the upstream driver of the PIK3-AKT-mTOR pathway. PI3K is commonly mutated in breast cancer, and activation of this pathway has been implicated in acquired and de novo resistance to endocrine therapy. There is crosstalk between the ER and PI3K/AKT pathways; however, they also signal independently, suggesting that dual pathway targeting may be required to optimize outcomes. The phase III trial, BELLE-2, is exploring the role of the PI3K inhibitor BKM120 with fulvestrant in women who have progressive ER+ MBC following an AI ( NCT01610284 ). An ongoing phase II trial ( NCT01437566 ) is studying fulvestrant with or without GDC0941, an alternative PI3K inhibitor in a similar population. This study is also examining the role of PI3K mutational status as a potential biomarker. The outcome of these studies will further inform the role of targeting this pathway.
HER2
In addition to mTOR inhibition, overexpression of the HER2 proto-oncogene has been clinically validated as a mediator of resistance to endocrine therapy. However, this applies to a small subgroup because only 10% of ER+ breast cancers are also HER2 positive. HER2-driven resistance to antiendocrine therapy is mediated via decreased ER level and increased ER phosphorylation, altered ER transcription, and activated downstream PI3K/AKT and MAPK pathways. Dual targeting of both ER and HER2 overcomes this resistance in preclinical models. Increased HER2 signaling is seen in 20% of HER2− patients relapsing on tamoxifen, although conversion to clinically HER2+ disease is rare. Crosstalk between the HER2 and ER pathways is thought to drive endocrine therapy resistance. In ER+ HER2− cell lines with acquired resistance, lapatinib, an oral tyrosine kinase inhibitor of HER2, was able to restore sensitivity. The phase III TAnDEM study randomized women with ER+ HER2+ MBC to anastrozole with or without trastuzumab, a monoclonal antibody to HER2. The combination improved response rate (RR) and PFS. A 4.6-month improvement in OS did not achieve statistical significance; however, 70% of patients on anastrozole crossed over to receive trastuzumab on disease progression. In another phase III study, lapatinib with letrozole improved response and PFS in women with ER+ HER2+ disease. There was no benefit with the addition of lapatinib to letrozole in women who had ER+ HER2− disease.
EGFR
EGFR contributes to endocrine resistance in preclinical models; however, clinical trials targeting this pathway have had only moderate success. A phase II study showed improved PFS with the addition of gefitinib to anastrozole as first-line therapy for ER+ MBC. Another phase II trial of gefitinib monotherapy showed a clinical benefit rate of 54% in tamoxifen-resistant MBC. Another phase II study of tamoxifen with gefitinib had a numerically improved PFS with the EGFR inhibitor, but there was no benefit in patients who had received prior endocrine therapy. In addition, a phase II study of anastrozole and the EGFR/HER2/HER3 inhibitor AZD8931 has been closed for futility. The role of EGFR inhibitors in this setting is therefore not well defined. Biomarkers to better predict response to EGFR inhibition may lead to appropriate patient selection for this targeted therapy.
HER2-positive breast cancer
The HER2 gene, also known as HER2/neu or c-erbB2, is located on chromosome 17q. HER2 belongs to the ErbB family of receptor tyrosine kinases, which normally regulate a series of cellular processes, including proliferation and growth. The HER2 gene is a proto-oncogene, a normal gene with the potential to become an oncogene as a result of molecular alterations, such as mutation, amplification, or overexpression of its protein product. The HER2 protein is overexpressed and/or its gene is amplified in approximately 20% of invasive breast cancers, and it is associated with a more aggressive biology, increased risk for progression of disease, and decreased OS. Advances in translational science have led to the development of several therapies that target HER2, including the monoclonal antibody trastuzumab and the small-molecule tyrosine kinase inhibitor (TKI) lapatinib; however, many tumors either exhibit de novo resistance to anti-HER2 therapy or acquire resistance over time, leading to disease progression and shortened survival for patients. More recently, novel agents with varying mechanisms of action have been described, and emerging data indicate that combinations of anti-HER2 agents may overcome resistance.
Trastuzumab
Trastuzumab is a humanized recombinant monoclonal antibody that binds to the external domain of HER2. On binding, trastuzumab downregulates the ligand-independent HER2 dimerization and growth factor signaling cascades downstream of HER2, including the PI3K/AKT/mTOR pathway. Trastuzumab mediates several antitumor mechanisms, including induction of an immune response to tumor through antibody-dependent cellular cytotoxicity, blockade of cleavage of the HER2 receptor, as well as downregulating ligand-independent HER2 dimers.
The clinical benefit of trastuzumab for patients with tumors that are HER2 positive (amplified or overexpressed) was first shown in 2001 in a pivotal phase III randomized trial in which the addition of trastuzumab to chemotherapy was associated with higher overall RRs, and improved PFS and OS, in patients with MBC. In 2005 trastuzumab was introduced in the adjuvant setting following publication of the results from several large phase III trials showing that the addition of 1 year of trastuzumab to postoperative chemotherapy decreased risk of recurrence by approximately one-half, and decreased risk of death from breast cancer by one-third. Longer follow-up from these studies has shown sustained benefits, and 1 year of therapy remains the standard of care. In the preoperative setting, the addition of trastuzumab to chemotherapy has been associated with an increase in the rates of pathologic complete response (pCR), from 22% to 44% in the NeOAdjuvant Heceptin Study and, similarly, from 16% to 32% in the phase III GeparQuattro Study, leading to the acceptance of chemotherapy plus trastuzumab as standard of care in the neoadjuvant setting for women with HER2-positive breast cancer.
The molecular basis for resistance to trastuzumab, either de novo or acquired, has been difficult to elucidate, in part because of the difficulty in obtaining tumor samples after tumor progression, and in part because trastuzumab has multiple modes of pharmacologic action. Several laboratory models of resistance to trastuzumab have been reported, including hyperactivation of the PI3K/AKT/mTOR pathway through loss of the phosphatidylinositol phosphate 3′-phosphatase (PTEN) tumor suppressor or mutational activation of PIK3CA, and alteration of HER2 expression either through overt loss of overexpression or through induction of a cleaved form of HER2 that lacks the extracellular domain. In a study of biopsy specimens from patients with tumors progressing on trastuzumab therapy, we found evidence for changes that activate PI3K/AKT signaling, but little evidence for loss of HER2 expression, suggesting that continued targeting of HER2 along with cotargeting of the PI3K/AKT pathway may represent a rational therapeutic approach.
Lapatinib
Lapatinib is an oral reversible, small-molecule dual inhibitor of both the EGFR (HER1) and HER2 kinases. In 2007 it was approved for use in trastuzumab-refractory HER2-positive MBC. Preclinical evidence suggests that lapatinib exerts its antitumor effects by inducing growth arrest and/or apoptosis, as well as by blocking downstream MAPK and AKT signaling pathways. Synergism for the combination of lapatinib and trastuzumab in HER2-positive disease has been shown in the metastatic setting and in the neoadjuvant setting by increased rates of pCR. The Adjuvant Lapatinib and/or Trastuzumab Treatment Optimization (ALTTO) study is expected to refine the treatment algorithm for HER2-positive breast cancer in the adjuvant setting.
Other Anti-HER2 Agents
Pertuzumab is a humanized monoclonal antibody that binds to an alternate extracellular domain of the HER2 receptor. Antibody binding prevents receptor dimerization and ligand-activated signaling with other growth factor receptors. Similar to lapatinib, recent studies have shown an improvement in PFS in the metastatic setting with the combination of pertuzumab and trastuzumab, and increased rates of pCR in the neoadjuvant setting.
The novel antibody drug conjugate, ado-trastuzumab emtansine (also called T-DM1), which couples trastuzumab with a chemotherapeutic agent with a microtubule binding effect similar to vinca alkaloids, was recently approved for HER2-positive metastatic disease, and a range of compounds to overcome resistance by targeting heat shock protein 90, a molecular chaperone required for the stabilization of cellular proteins, are under development.
In addition, as described earlier, possible mechanisms of resistance to trastuzumab include loss of PTEN and/or activation of the PI3K/AKT signaling pathway, and these observations have led to investigation of the role of mTOR, which is a downstream component of the PTEN/PI3K pathway. As such, mTOR inhibitors, such as everolimus, are under investigation for HER2-positive MBC.
Related posts:

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


