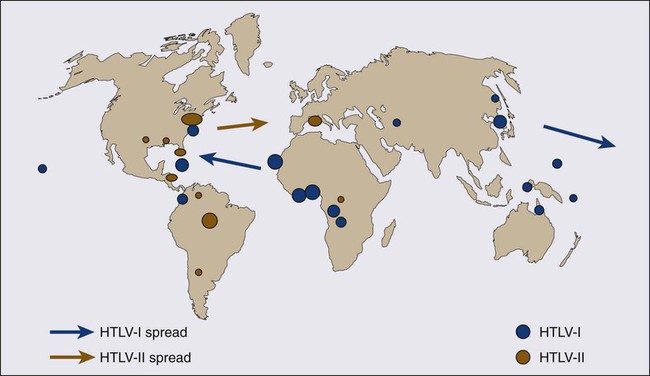
Kunihiro Tsukasaki, Toshiki Watanabe and Kensei Tobinai • Adult T-cell leukemia-lymphoma (ATL) is a distinct peripheral T-cell malignancy associated with human T-cell leukemia/lymphotropic virus type I (HTLV-I). • HTLV-I is reverse-transcribed into DNA and randomly integrated into the host cell. • The HTLV-I genome encodes two unique regulatory proteins—Tax and Rex—responsible for viral expression and cellular transformation. Tax trans-activates viral and cellular genes that could be involved in the pathogenesis of ATL. HTLV-I basic leucine zipper (HTLV-I bZIP; HBZ) is an antisense transcript of HTLV-I, is steadily expressed in ATL cells, and interacts with several host genes and suppresses the activity of Tax. • A major cluster of HTLV-I–infected individuals and patients with ATL exists on the southwest coast of Japan, where approximately 1.1 million people are infected with the virus. • Other clusters have been noted in the Caribbean islands (African), tropical Africa (African), South America (Mongoloid), and northern Oceania (Melanesian). • HTLV-I is transmitted from mother to child through breast-feeding, by sexual contact, and by blood-borne transmission. • The estimated cumulative risk of the development of ATL in HTLV-I–positive individuals is about 3% after transmission from their mothers. • Patients with ATL show diverse clinical features, and four clinical subtypes have been recognized: acute, lymphoma, chronic, and smoldering. • The typical manifestations of acute-type ATL include circulating neoplastic cells in the peripheral blood, generalized lymph node swelling, hepatosplenomegaly, skin involvement, opportunistic infections, and hypercalcemia. • Leukemic cells in the peripheral blood characteristically show markedly polylobated nuclei, so-called flower cells. Their immunophenotypes are CD4/CD25/CCR4+ and CD8− in most cases. • All histopathological specimens show findings of peripheral T-cell lymphoma of various subtypes. • ATL is suspected when the aforementioned characteristic clinical manifestations and/or the cytologic findings of leukemic cells in the peripheral blood are recognized. • Morphologic and immunophenotypic analyses of neoplastic cells in peripheral blood or tumor lesions and a serologic assay against HTLV-I are required for the clinical diagnosis of ATL. • The demonstration of the monoclonal integration of HTLV-I proviral DNA in the neoplastic cells can lead to a definite diagnosis of ATL. • An accurate diagnosis of the clinical subtype is vital for appropriate decisions regarding treatment. • Intensive chemotherapies combining agents used in the treatment of non-Hodgkin lymphoma (NHL) are usually given to patients with the acute or lymphoma subtype of ATL; however, most patients with ATL do not achieve cure with current chemotherapy regimens. • Further efforts to incorporate promising or new, innovative treatment modalities, such as interferon/zidovudine therapy, new anticancer agents, monoclonal antibody therapy, molecular-targeting therapy, and allogeneic hematopoietic stem cell transplantation (allo-HSCT), are needed for the establishment of risk-adopted therapy. • Prevention of HTLV-I infections has been achieved in some endemic areas by screening for HTLV-I among blood donors and recommending mothers who are carriers to refrain from breast-feeding. Prevention of ATL among HTLV-I carriers has not been achieved, although several risk factors have been identified. Adult T-cell leukemia-lymphoma (ATL) was first recognized in Japan in 1977.1 The disease was characterized as leukemia of peripheral T cells, generalized lymphadenopathy, hepatosplenomegaly, and skin involvement. Because of its unusual geographic clustering in southwestern Japan, it was postulated that some infectious agent(s) had causative roles. Human T-cell leukemia/lymphotropic virus type I (HTLV-I) was first isolated by Poiesz and associates2 in the United States from cultured cells from one patient with an aggressive variant of mycosis fungoides and from one with Sézary syndrome. Although both patients, who were African Americans, were diagnosed clinically as having cutaneous T-cell lymphoma (CTCL) at the time of reporting, their clinical features were later found to closely resemble those of Japanese patients with ATL. In 1980, Miyoshi and coworkers3 established the first cell line (MT-1) derived from neoplastic cells in an ATL patient. They co-cultured neoplastic cells from an ATL patient with normal human cord blood leukocytes and established the cell line MT-2 (derived from cord blood lymphocytes), which produced large amounts of type C retrovirus.4 Using the MT-1 cell line, Hinuma and colleagues5 found that patients with ATL had antibodies against the virus-associated antigen in their sera. The “ATL virus” was then isolated and characterized as an RNA retrovirus.6 Because the ATL virus was found to be identical to HTLV by a DNA sequence analysis, it was designated HTLV-I.7 The etiologic association of HTLV-I and ATL is based on the following findings: • The areas with a high incidence of patients with ATL closely correspond to those with a high prevalence of HTLV-I carriers.8 • HTLV-I immortalizes T cells in vitro.9 • HTLV-I proviral DNA is detected in the neoplastic cells of patients with ATL.10 • Almost all patients with ATL have antibodies against HTLV-I in their sera. HTLV-I is the first retrovirus found to be associated with a malignant neoplasm in humans. The RNA genome of HTLV-I is reverse-transcribed into DNA and integrated as a proviral DNA into the host cell. The HTLV-I provirus is 9.0 kilobases long and has structural genes in the order 5′-gag-pol-env-3′. Both ends of the HTLV-I proviral DNA contain repeats called long terminal repeats (LTRs). The HTLV-I gene encodes three structural proteins: group antigen (gag), reverse transcriptase (pol), and envelope (env) proteins. The full-length mRNA is used for the synthesis of gag and pol gene products. The gag protein is synthesized as a precursor polypeptide of 55 kDa that is proteolytically cleaved into the individual gag proteins p19, p24, and p15. The protease is encoded in a different reading frame that spans the 3′ part of the gag region and the 5′ part of the pol region. The pol region encodes the reverse transcriptase, integrase, and RNase H. The env gene encodes two proteins made from a singly spliced mRNA. It is then cleaved intracellularly into an extracellular glycosylated protein (gp46) and a transmembrane (gp21). The pX region at the 3′ end of the genome has the potential to encode essential regulatory proteins (Tax and Rex) and accessory proteins—p8, p12, p13, and p30—that are important for viral infectivity and replication by influencing cellular signaling and gene expression.11–16 The life cycle of a retrovirus begins with the binding of the virus to specific receptors on the cell surface via viral envelope proteins. Three molecules, glucose transporter–1 (GLUT1), neuropilin-1 (NRP1), and heparan sulfate proteoglycans (HSPG) are involved in HTLV-I binding and entry.17–21 HTLV-I transmission is mediated by cell-to-cell contact. A unique structure called the “virologic synapse” was reported to form at the contact site of HTLV-I–infected cells and uninfected T cells, and viral materials are transmitted through it. On the other hand, it was reported that HTLV-I particles move from cell to cell in a complex with numerous extracellular matrix components, forming large surface-associated biofilm-like structures.22 In addition, HTLV-I p12/p8 expression increases T-cell contact through specific adhesion molecules and promotion of cellular conduits appears to enhance cell-to-cell viral transmission.16 The onset of ATL is preceded by a long period of clinical latency, frequently lasting more than 4 decades. In addition, ATL develops in less than 5% of all individuals infected with HTLV-I. The promoter insertion model was rejected as the leukemogenic mechanism because integration sites of the provirus were random, depending on the patient.11 Consequently, a trans-acting viral factor, Tax, has been shown to be oncogenic, because it transforms and immortalizes rodent fibroblasts and T lymphocytes as well as human T lymphocytes. Tax trans-activates viral transcription through interaction with the cellular basic domain/leucine zipper transcription factors cyclic adenosine monophosphate (cAMP) response element–binding (CREB) factor and cAMP-dependent transcription factor–1 (ATF1). Tax interacts with numerous cellular proteins to reprogram cellular processes, including, but not limited to, transcription, cell cycle regulation, DNA repair, and apoptosis. Tax transcriptionally regulates cellular genes by interaction with enhancer-binding proteins such as CREB, nuclear factor–κB (NF-κB), and serum response factor and by tethering coactivators to the DNA-bound transcription factors. Tax also stimulates cell growth by direct binding to cyclin-dependent kinase holoenzymes and/or inactivating tumor suppressors such as p 53 (tumor protein p53) and DLG1 (discs, large). Furthermore, Tax silences cellular checkpoints, which guard against DNA structural damage and chromosomal missegregation, thereby favoring the manifestation of a mutator phenotype in cells.13,23 Tax interacts and activates specific components of growth factor signal transduction pathways, such as IκB kinase–nuclear factor–κB (IκB kinase [IKK]-NF-κB), RAS/mitogen-activated protein kinase, protein kinase A, and protein kinase C.24,25 Interaction with IKKγ, a component of the IKK complex, results in the constitutive activation of this kinase complex.26 Constitutive activation of the Janus-activated kinase–signal transducer and activator of transcription (JAK-STAT) pathway in HTLV-I–transformed cells has also been reported, although the mechanisms are not well understood.27 Thus, HTLV-I infection results in the aberrant activation of growth-promoting signaling pathways. The oncogenic capacity of Tax has been reported in various systems; however, cellular transformation by HTLV-I in vivo is a multistage process and viral gene expression is absent in ATL cells in vivo.28,29 Moreover, proviruses integrated in ATL cells are frequently defective, have mutations in the coding region of Tax, and/or are methylated in the 5′ and 5′ LTR regions.32–32 Thus, in addition to promoting growth directly, Tax should endow the infected T cells with capacities that aid the progression to transformed phenotypes in the absence of Tax. In this context, induction of a mutator phenotype by Tax in the infected cells appears to play an important role.33 The role of HTLV-I Tax in the multiple-step leukemogenesis of ATL are illustrated in Figure 108-1. The expression of antisense strand RNA with the capacity to encode a zinc finger protein (HTLV-I basic leucine zipper [HTLV-I bZIP; HBZ] factor) has opened up a new field of research. HBZ inhibits Tax-dependent viral transcription.34 HBZ RNA is transcribed from the region of the HTLV-I provirus that is spared genomic deletion and has been shown to be expressed in vivo in all ATL patients as well as virus carriers; thus, it might be involved in the growth of ATL cells.35 Various karyotypic abnormalities have been reported in neoplastic cells of ATL; however, no specific karyotypic abnormality has been found. In general, the chromosomal abnormalities are more complex in the acute type than in the chronic type. Itoyama and colleagues36 reported the results of cytogenetic analysis of 50 cases of ATL and found aneuploidy and multiple breaks more frequently in acute and lymphoma types. Multiple breaks and partial loss of chromosomes correlated with shorter survival. The authors claim that one model of an oncogenic mechanism—activation of a proto-oncogene by translocation of a T-cell receptor (TCR) gene—might not be applicable to the main pathway of development of ATL and that a multiple-step process of leukemogenesis is required. In a study by Tsukasaki and colleagues,37 64 patients with ATL were analyzed by using comparative genomic hybridization (CGH). The most frequent observations were gains at 14q, 7q, and 3p and losses at 6q and 13q. Chromosomal imbalances, losses, and gains were observed more frequently in acute or lymphoma types. An increased number of chromosomal imbalances was associated with a shorter survival. Paired samples (i.e., samples obtained at different sites) from 4 patients and sequential samples from 13 patients (from 6 during both the chronic phase and acute crisis and from 7 during both acute onset and relapse) were examined by CGH and Southern blotting for HTLV-I. All but two paired samples showed differences on CGH assessment. Two chronic/crisis samples showed distinct results regarding both CGH and HTLV-I integration sites, suggesting clonal changes in ATL at crisis. In 11 patients, the finding of identical HTLV-I sites and clonally related CGH results suggested a common origin of sequential samples. In contrast to chronic/crisis samples, CGH results with all acute/relapse sample pairs showed the presence of clonally related but not evolutional subclones at relapse. It was concluded that clonal diversity is common during the progression of ATL and that CGH alterations are associated with clinical course. P53 is a nuclear phosphoprotein that functions as a tumor suppressor. A loss of a normally functioning p53 through mutation or allelic loss has been found in several kinds of malignant neoplasms. Mutations of the p53 gene have also been found in some patients with ATL.38,39 According to a study by Cesarman and coworkers,39 no p53 mutations were detected in samples from 11 patients with the chronic type of ATL, whereas 9 (28%) of 28 samples from patients with the acute type exhibited p53 mutations. In one patient, a tumor sample obtained during the chronic phase did not have a mutation of p53 but the mutation was subsequently detected in a sample that was obtained at crisis. These results suggest that alterations of the p53 gene might contribute to disease progression in a fraction of patients with ATL. Other putative tumor suppressor genes, p15INK4B and p16INK4A, were reported to be associated with ATL.42–42 Yamada and colleagues41 reported that 28 (25%) of 114 patients with ATL showed homozygous deletions of the p15 and/or p16 genes. These results correlated well with the clinical subtype of ATL. In addition, the patients with deleted p15 and/or p16 genes had significantly shorter survival period than did patients in whom both genes were preserved (P < .0001). Moreover, three of the five patients with chronic-type ATL who progressed to acute-type ATL lost the p16 gene alone or both genes at their exacerbation phase. These results suggest the deletion of p15 and/or p16 to play a key role in the disease progression of some patients with ATL. Uchida and colleagues42 found a point mutation of the p16 gene in 3 (7%) of 44 patients with ATL. It is suggested that the p16 gene is inactivated not only by homozygous deletion but also by point mutation. Several investigators have analyzed the implications of the integration pattern of HTLV-I provirus in the progression of ATL.43,44 It is known that the neoplastic cells of ATL have one complete copy of the HTLV-I provirus per cell in some patients (complete type) whereas others have multiple complete copies per cell (multiple type). The HTLV-I proviruses in the remaining patients do not have the complete genome but rather have a defective genome (defective type). Tsukasaki and colleagues44 found that the median survival times (MSTs) for patients were 7 months, 24 months, and 33 months for defective-type, complete-type, and multiple-type ATL, respectively (P = .006). Among 52 sequentially examined patients, the HTLV-I integration patterns changed in four patients (8%). In three of these four, the rearrangements of the TCRβ gene changed concomitantly, suggesting the appearance of a new ATL clone. The researchers concluded that the frequent clonal change of ATL at crisis reflects the emergence of multiple premalignant clones in viral leukemogenesis. Tamiya and coworkers43 reported the presence of two types of defective viruses. The type 2 defective virus with a deletion that includes the 5′ LTR was found more frequently in the acute and lymphoma types (39%, 21 of 54) than in the chronic type (6%, 1 of 18). It is postulated that the high frequency of the type 2 defective virus is caused by the genetic instability of the HTLV-I provirus and that this defective virus is selected for because it evades the host’s immune surveillance system. HTLV-I is an etiologic agent not only in ATL but also in the neurologic disorder known as tropical spastic paraparesis (TSP) or HTLV-I–associated myelopathy (HAM).45,46 In TSP/HAM the HTLV-I provirus remains randomly integrated, whereas in ATL the provirus is monoclonally integrated. Inhabitants of the southwest coast of Japan have the highest prevalence of HTLV-I infections and the highest incidence of ATL in the world.8,46–48 A high prevalence of HTLV-I is also found in inhabitants of the Caribbean islands (African), tropical Africa (African), South America (Mongoloid), and northern Oceania (Melanesian).47–50 Many patients who have been diagnosed with ATL in Western countries are immigrants from the West Indies and tropical Africa. The world map of the distribution of HTLV-I and HTLV-II and the presumed routes of spread is shown in Figure 108-2.48 The geographic clustering of HTLV-I carriers is suggested to be strongly associated with high frequencies of mother-to-child and sexual transmissions of the virus under closed conditions in particular groups.51 Recently, the prevalence of HTLV-I in Japan as determined by screening of blood donors was surveyed.52 The seroprevalence of HTLV-I among 1,196,321 Japanese first-time blood donors from 2006 to 2007 was investigated. A total of 3787 such donors were confirmed to be positive for the anti–HTLV-I antibody. By applying a fitness curve to the age ranges outside the blood donor age range, the present number of HTLV-I carriers covering ages from birth to 99 years was estimated to be at least 1.08 million in Japan; this value was 10% lower than that reported in 1988.53 The adjusted overall prevalence rates were estimated to be 0.66% and 1.02% in men and women, respectively. The peak in carrier numbers was found among individuals in their 70s, which is a shift from the previous peak observed in the 1988 database among individuals in their 50s. As compared with the survey in the 1980s, carriers were distributed not only in the endemic southwestern region of Japan but also throughout the country, particularly in the greater Tokyo metropolitan area. The authors calculated, after applying population projections, that the carrier number will decrease by half in the next two decades; however, the carrier population will age over that interval, meaning that the age of patients with ATL will also be higher. In endemic areas, there is a marked increase in HTLV-I prevalence with age until 70 years and an increased prevalence among females compared with males. Transmission occurs via sexual and blood-borne routes. A major reason for the increase in seroprevalence with age appears to be the decreasing prevalence of HTLV-I in the population over time, at least in Japan, where it has been most extensively studied. Yamaguchi and coworkers54 reported that the HTLV-I carrier rates among blood donors at Kyusyu in Japan had fallen since 1986 in all age groups younger than age 50 years and in both genders. Recently, the results were reconfirmed in another endemic area in Japan.55 This decrease in HTLV-I carriers among younger blood donors might be explained by improvements in sanitation and general lifestyle changes in recent decades. A shorter duration of breast-feeding, the increasing use of artificial feeding for infants, and decreasing family sizes are also likely to be factors for the recent decline in the vertical transmission rates of HTLV-I.54,55 Overall, there is a slight male predominance of ATL patients, with the male-to-female ratio ranging from 1.1 to 1.5. This ratio contrasts with that of TSP/HAM, which affects females more frequently than males. 1. Mother-to-child-transmission, mainly by HTLV-I–positive lymphocytes in breast milk56 2. Sexual transmission, more commonly from males to females 3. Blood-borne transmission, including blood transfusions and sharing of needles by intravenous drug abusers57,58 The first infection route is vertical transmission from mother to child via HTLV-I–positive lymphocytes in breast milk. The overall infection rate for HTLV-I in children with seropositive mothers has been estimated to be 10% to 30%. HTLV-I-infection has also been reported in about 3% of children not breast-fed, which suggests the possibility of an intrauterine or transvaginal infection.59 However, the intrauterine route was unlikely, given the discordance of HTLV-I DNA in cord blood and the subsequent seroconversion of the infants.60 Several types of intervention have been conducted in HTLV-I–endemic areas in Japan, where seropositive pregnant women are advised not to breast-feed.51 Recently, a nationwide intervention has been initiated in Japan. The second route is transmission through sexual contact. Transmission of HTLV-I frequently occurs from males to females but rarely from females to males, similar to human immunodeficiency virus (HIV) transmission. HTLV-I has been isolated in semen. It appears likely that the risk of development of ATL after HTLV-I infection by this route of transmission is not high because most of the mothers of ATL patients were seropositive for HTLV-I.61 To prevent HTLV-I transmission through blood transfusions, serologic screening of all blood donors for HTLV-I has been conducted in Japan since November 1986. The present donor screening program for HTLV-I has almost completely prevented transmission by transfusion in Japan. In contrast to red cell and platelet concentrates, fresh-frozen plasma and plasma fractions have never been shown to transmit HTLV-I.57 Approximately 1.1 million HTLV-I–infected individuals reside in Japan, and the annual incidence of ATL in Japan is approximately 1000.52 The annual rate of ATL development among HTLV-I carriers older than 40 years is estimated at 1.5 per 1000 in males and 0.5 per 1000 in females, and the cumulative risk of ATL development among the HTLV-I carriers is estimated to be 2.5% to 5% over the course of a 70-year life span.62 It was postulated that the leukemogenesis of ATL consists of five steps, based on a statistical analysis of ATL development according to age.63 A recent nationwide survey in Japan revealed that compared with previous nationwide studies, the age of ATL patients shifted toward older ages and the mean age gradually increased from 52.7 years in the first survey (cases before 1980) to 61.1 years in the ninth survey (1996 to 1997) and, finally, to 66.0 years in the 10th study (range, 19 to 94; median, 67).64 It has been reported that the age of patients with ATL in areas outside Japan is lower, with an overall mean age in the mid 40s.65 HTLV-I infection early in life, presumably from breast-feeding, is crucial in the development of ATL; 100% of mothers of patients with ATL were HTLV-I carriers as compared with about 30% of mothers of patients with TSP/HAM.61 HTLV-I infection by blood transfusion is associated with a higher risk for the development of TSP/HAM than infection by other routes.66 In contrast, very few cases of ATL after HTLV-I infection by blood transfusion have been reported.67,68 Interestingly, those affected had blood transfusions for a preceding hematologic malignancy and were diagnosed with ATL within 11 years after the HTLV-I infection. Recently, the development of ATL after kidney, living donor liver, and hematopoietic stem cell transplantations has been reported, possibly associated with the use of immune suppressants.71–71 Other factors reportedly associated with the onset of ATL include HTLV-I infection early in life, increase in age, male sex, family history of ATL, past history of infective dermatitis, smoking of tobacco, serum titers of antibody against HTLV-I, and several human leukocyte antigen (HLA) subtypes.72–77 However, definitive risk factors for the development of ATL among asymptomatic HTLV-I carriers have not been determined. HTLV-I proviral loads have been proposed as an important predictor of the development of ATL, but only a few small prospective studies have been conducted. Recently, Iwanaga and colleagues evaluated 1218 asymptomatic HTLV-I carriers (426 males and 792 females) who were enrolled during 2002 through 2008 for a prospective study on the development of ATL.78 The proviral load at enrollment was significantly higher in males than females (median, 2.10 vs. 1.39 copies/100 peripheral blood mononuclear cells (PBMCs) (P < .0001), in those aged 40 or older, and in those with a family history of ATL. During the follow-up period, 14 participants developed acute ATL. Their baseline proviral loads were high (range, 4.17 to 28.58 copies/100 PBMCs). Multivariate Cox regression analyses indicated that not only a higher proviral load but also advanced age, a family history of ATL, and the first opportunity for HTLV-I testing during treatment for other diseases were independent risk factors for the progression of ATL from a carrier status. • Viral infection alone is not sufficient for the expression of the malignant phenotype. • The timing and/or length of viral exposure are critical. • The long latency period suggests that the disease progression is a multiple-step process. This contrasts with TSP/HAM, which can occur with a shorter latency period, especially among recipients of blood transfusions. After HTLV-I was revealed to be associated with ATL, it was found that ATL shows a marked diversity in its clinical manifestations. ATL cases have been subdivided into four distinct clinicopathological entities: acute, lymphoma, chronic, and smoldering types. The recognition of the four clinical subtypes is important in understanding the natural history, clinical features, treatment strategy, and leukemogenesis of ATL. On the basis of a nationwide survey of 854 patients with ATL who were diagnosed between 1983 and 1987 in Japan, the Lymphoma Study Group proposed diagnostic criteria for the four clinical subtypes based on the sites of organ infiltration, presence, absence and degree of leukemic manifestation, a high lactic acid dehydrogenase (LDH) value, and hypercalcemia (Table 108-1)79: Table 108-1 Diagnostic Criteria for Clinical Subtypes of Adult T-Cell Leukemia-Lymphoma HTLV-I, Human T-cell leukemia/lymphotropic virus I; N, normal upper limit. *No essential qualification except terms required for other subtype(s). †Typical “flower cells” may be seen occasionally. ‡Accompanied by T lymphocytosis (3.5 × 103/µL or more). §If the proportion of abnormal T lymphocytes is less than 5% in peripheral blood, a histologically proven tumor lesion is required. ¶Histologically proven skin and/or pulmonary lesion(s) is required if there are fewer than 5% abnormal T lymphocytes in peripheral blood. From Shimoyama M and Members of the Lymphoma Study Group (1984-1987). Diagnostic criteria and classification of clinical subtypes of adult T-cell leukemia-lymphoma. Br J Haematol 1991;79:428. 1. The acute type shows a rapidly progressive clinical course and most of the characteristic features of ATL: leukemic manifestation, generalized lymphadenopathy, hepatomegaly, splenomegaly, skin involvement, hypercalcemia, and the infiltration of other organs (central nervous system, gastrointestinal tract, etc.). The symptoms and signs include abdominal pain, diarrhea, ascites, jaundice, pleural effusion, cough, sputum, fever, and unconsciousness because of organ involvement, hypercalcemia, and/or opportunistic infections. 2. The smoldering type shows an indolent clinical course and only a small percentage of leukemic cells, but it also can include skin and lung involvement. 3. The chronic type, lymphocytosis with a high percentage of leukemic cells, is occasionally associated with skin and lung involvement, lymphadenopathy, and hepatosplenomegaly and also shows an indolent clinical course. 4. The lymphoma type includes patients who have the manifestations of nodal NHL without circulating malignant cells in the peripheral blood. ATL, particularly the aggressive forms (acute and lymphoma types), has been found to infiltrate the stomach and the intestines in 29% and 25% of patients, respectively, at autopsy.80 The involvement may be focal as an isolated gastric lesion or so diffuse as to involve the entire gastrointestinal tract. Extensive infiltration of the intestines can lead to moderate to severe diarrhea and malabsorption. Patients with ATL suffer from a variety of abdominal symptoms (e.g., nausea, vomiting, abdominal fullness, and diarrhea), which might be attributable to infiltration by neoplastic cells, but, because of the associated immunodeficiency, various opportunistic infections such as strongyloidiasis and cytomegalovirus (CMV) gastrocolitis can complicate cases. Hepatic involvement of ATL cells can be found in up to one fourth of patients with acute and lymphoma subtypes and not infrequently manifests as jaundice and elevated hepatic transaminase levels. Among patients with acute-type or lymphoma-type ATL, palpable hepatomegaly was more frequent and total bilirubin, hepatic transaminase, LDH, and alkaline phosphatase values were higher than among other patients with NHL. Autopsy liver samples disclosed that the portal area was most frequently infiltrated with ATL cells.81 Pulmonary complications, which are common in ATL, are due to leukemic infiltration in one half of patients and to infections with a variety of bacterial and opportunistic organisms in the other half.82 Of 854 Japanese patients with ATL, 26% had active infections at the time of diagnosis.78 The incidence was highest among patients with the chronic and smoldering types (36%) and lower for patients with the acute (27%) and lymphoma (11%) subtypes. The infections that were encountered were bacterial (pneumonia, sepsis, and tuberculosis) in 43%, fungal in 31%, protozoal in 18%, and viral in 8% of patients with ATL (Table 108-2). The immunodeficiency at presentation in ATL can be exacerbated by the neutropenia that is produced by cytotoxic chemotherapy, leading to an extremely high risk of infection throughout the course of therapy. Infections are responsible for the patient’s death in about half of the cases. Pulmonary involvement of ATL cells occurred in 17% of the patients.79 Table 108-2 Infectious Complications at Diagnosis in 818 Japanese Patients with Adult T-Cell Leukemia-Lymphoma *Values in parentheses indicate the total number of patients in each category. ‡One patient each suffered from oral candidiasis. From Shimoyama M and Members of the Lymphoma Study Group (1984-1987). Diagnostic criteria and classification of clinical subtypes of adult T-cell leukemia-lymphoma. Br J Haematol 1991;79:42. Central nervous system (CNS) involvement occurs in approximately 10% of patients with ATL.83 Opportunistic infections of CNS such as cryptococcal meningitis and toxoplasmosis and hypercalcemia can also cause CNS symptoms and signs in ATL patients. Cutaneous involvement occurs in approximately 40% of patients with ATL. The incidence was highest among patients with the chronic and smoldering types (48%) and lower for patients with the acute (40%) and lymphoma (25%) subtypes.79 A proportion of patients with smoldering ATL have cutaneous involvement without leukemic manifestation.84 Although cutaneous involvement is relatively frequently observed in indolent (smoldering or chronic) ATL, the primary cutaneous tumoral type, although generally included among smoldering ATL, was reported as having a poor prognosis.85,86
Adult T-Cell Leukemia-Lymphoma
Introduction
Virology and Pathogenesis
Role of Tax
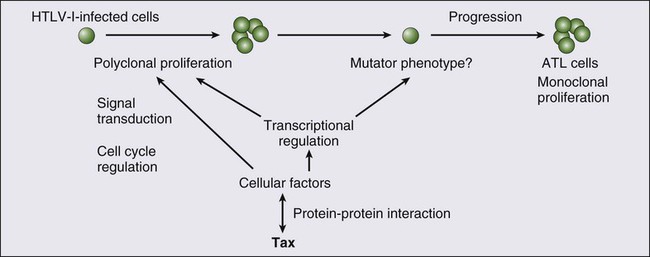
Presence of Antisense Transcript and Its Function
Role of Chromosomal Abnormalities
Role of p53 and Other Tumor Suppressor Genes
Role of HTLV-I Provirus
Epidemiology of HTLV-I and Adult T-Cell Leukemia-Lymphoma
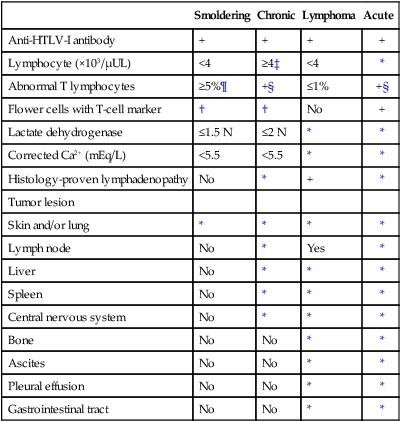
Clinical Manifestations
Smoldering
Chronic
Lymphoma
Acute
Anti-HTLV-I antibody
+
+
+
+
Lymphocyte (×103/µUL)
<4
≥4‡
<4
*
Abnormal T lymphocytes
≥5%¶
+§
≤1%
+§
Flower cells with T-cell marker
†
†
No
+
Lactate dehydrogenase
≤1.5 N
≤2 N
*
*
Corrected Ca2+ (mEq/L)
<5.5
<5.5
*
*
Histology-proven lymphadenopathy
No
*
+
*
Tumor lesion
Skin and/or lung
*
*
*
*
Lymph node
No
*
Yes
*
Liver
No
*
*
*
Spleen
No
*
*
*
Central nervous system
No
*
*
*
Bone
No
No
*
*
Ascites
No
No
*
*
Pleural effusion
No
No
*
*
Gastrointestinal tract
No
No
*
*
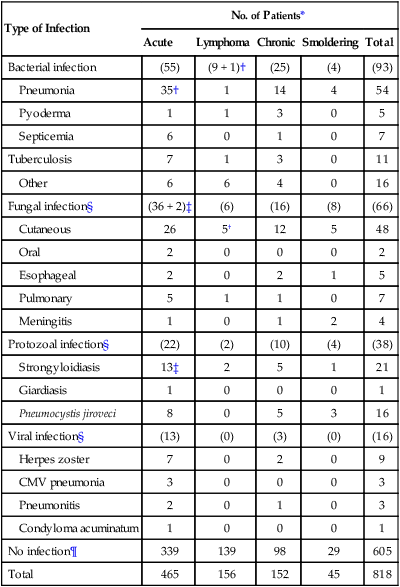
Type of Infection
No. of Patients*
Acute
Lymphoma
Chronic
Smoldering
Total
Bacterial infection
(55)
(9 + 1)†
(25)
(4)
(93)
Pneumonia
35†
1
14
4
54
Pyoderma
1
1
3
0
5
Septicemia
6
0
1
0
7
Tuberculosis
7
1
3
0
11
Other
6
6
4
0
16
Fungal infection§
(36 + 2)‡
(6)
(16)
(8)
(66)
Cutaneous
26
5†
12
5
48
Oral
2
0
0
0
2
Esophageal
2
0
2
1
5
Pulmonary
5
1
1
0
7
Meningitis
1
0
1
2
4
Protozoal infection§
(22)
(2)
(10)
(4)
(38)
Strongyloidiasis
13‡
2
5
1
21
Giardiasis
1
0
0
0
1
Pneumocystis jiroveci
8
0
5
3
16
Viral infection§
(13)
(0)
(3)
(0)
(16)
Herpes zoster
7
0
2
0
9
CMV pneumonia
3
0
0
0
3
Pneumonitis
2
0
1
0
3
Condyloma acuminatum
1
0
0
0
1
No infection¶
339
139
98
29
605
Total
465
156
152
45
818
Oncohema Key
Fastest Oncology & Hematology Insight Engine