Abbreviations
- ACE Angiotensin-converting enzyme
- ACTH Adrenocorticotropic hormone
- AM Adrenomedullin
- APMO Adnexal cystadenomas of probable mesonephric origin
- ARDS Acute respiratory distress syndrome
- ATP Adenosine triphosphate
- cAMP Cyclic adenosine monophosphate
- CgA Chromogramin A
- CGRP Calcitonin gene–related peptide
- COMT Catecholamine-O-methyltransferase
- DBH Dopamine-β-hydroxylase
- FDG Fluorodeoxyglucose
- DHMA Dihydroxymandelic acid
- DHPG Dihydroxyphenylglycol
- ECD Electrochemical detection
- HIF Hypoxia-inducible factor
- HPLC High-pressure liquid chromatography
- IP3 Inositol triphosphate
- MAO Monoamine oxidase
- MEN Multiple endocrine neoplasia
- MHBA 3-methoxy-4-hydroxybenzylamine
- MIBG Metaiodobenzylguanidine
- NF-1 Neurofibromatosis type 1
- NSE Neuron-specific enolase
- PBSC Peripheral blood stem cell
- PNMT Phenylethanolamine- N-methyltransferase
- PTHrP Parathyroid hormone–related peptide
- RECIST Response evaluation criteria in solid tumors
- SDH Succinate dehydrogenase
- SDHB Succinate dehydrogenase subunit B
- SDHC Succinate dehydrogenase subunit C
- SDHD Succinate dehydrogenase subunit D
- SPECT Single photon emission computed tomography
- SRI Somatostatin receptor imaging
- VEGF Vascular endothelial growth factor
- VHL Von Hippel-Lindau
- VIP Vasoactive intestinal polypeptide
- VMA Vanillylmandelic acid (3-methoxy-4-hydroxymandelic acid)
Adrenal Medulla and Paraganglia: Introduction
The adrenal medulla and paraganglia are part of the autonomic/sympathetic nervous system. The endocrine and nervous systems are alike in that they exert their actions by releasing hormones/neurotransmitters that bind to cell surface receptors in the target tissue, thereby inducing an effect.
Autonomic nerves are not under conscious control. They innervate the heart, adrenal medulla, vascular smooth muscle, and smooth muscle in visceral organs, thereby controlling cardiac rate and output, adrenal medullary secretion of catecholamines, blood pressure, the genitourinary tract, and intestinal motility. Autonomic nerves originate within the central nervous system and have two major divisions according to their anatomic locations:
- (1) Parasympathetic preganglionic nerves exit the central nervous system via the cranial nerves and sacral spinal nerves. They terminate in nonchromaffin paraganglia that are most numerous in the neck and associated with the glossopharyngeal and vagus nerves. These ganglia serve as chemoreceptors that are involved in the control of respiration. An important paraganglion at the carotid bifurcation is known as the carotid body. They are also found along the jugular vein and in the jugulo-tympanic region. Head-neck paragangliomas are tumors that arise from these parasympathetic paraganglia.
- (2) Sympathetic preganglionic nerves exit the central nervous system via the thoracic and lumbar spinal nerves. The sympathetic nervous system coordinates the body’s automatic fight-flight response by stimulating the adrenal medulla to secrete catecholamines and by directly stimulating cardiac output and blood flow to muscles while diverting blood flow away from visceral organs.
Sympathetic preganglionic nerves terminate mainly in paravertebral and prevertebral nerve ganglia where they secrete acetylcholine as their neurotransmitter; they are, therefore, known as cholinergic nerves. These nerve ganglia are collectively known as paraganglia and contain neuroendocrine cells that are similar to adrenal medullary cells on light microscopy by chromaffin and immunohistochemical staining. Paraganglia are also found in the mediastinum, particularly adjacent to the cardiac atria, and in the abdomen along the sympathetic nerve chains in paravertebral and prevertebral positions. Paraganglia are plentiful along the aorta, particularly around the celiac axis, adrenal glands, renal medullae, and aortic bifurcation (organ of Zuckerkandl). Paraganglia are also abundant in the pelvis, particularly adjacent to the bladder. Preganglionic nerves also terminate in the adrenal medulla, which is basically a sympathetic ganglion that is surrounded by adrenal cortex.
Sympathetic postganglionic nerve fibers originate from the paraganglia and run to the tissues being innervated. They secrete norepinephrine as their neurotransmitter at synaptic junctions. Adrenal medullary cells are basically modified postganglionic nerves that lack axons and secrete their neurotransmitter (mainly epinephrine) directly into the blood; thus, the bloodstream acts like a giant synapse, carrying epinephrine to receptors throughout the body. Synonyms for epinephrine and norepinephrine are adrenalin and noradrenaline, respectively (see later).
Although the adrenal medulla is not essential for survival, its secretion of epinephrine and other compounds helps maintain the body’s homeostasis during stress. Investigations of the adrenal medulla and the sympathetic nervous system have led to the discovery of different catecholamine receptors and the production of a wide variety of sympathetic agonists and antagonists with diverse clinical applications.
Pheochromocytomas are tumors that arise from the adrenal medulla, whereas non–head-neck paragangliomas arise from extra-adrenal sympathetic ganglia. Pheochromocytomas can secrete excessive amounts of both epinephrine and norepinephrine, whereas paragangliomas secrete only norepinephrine. The excessive secretion of catecholamines can result in a dangerous exaggeration of the stress response.
Figure 11–1
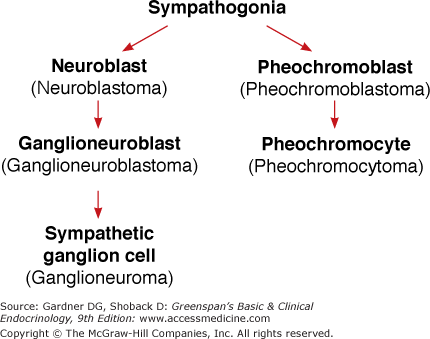
The embryonic development of adrenergic cells and tumors that develop from them (in parentheses). Sympathogonia are primitive cells derived from the neural crest. Neuroblasts are also called sympathoblasts; ganglion cells are the same as sympathocytes; and pheochromocytes are mature chromaffin cells.
The sympathetic nervous system arises in the fetus from the primitive cells of the neural crest (sympathogonia). At about the fifth week of gestation, these cells migrate from the spinal ganglia in the thoracic region to form the sympathetic chain posterior to the dorsal aorta. They then begin to migrate anteriorly to form the remaining ganglia.
At 6 weeks of gestation, groups of these primitive cells migrate along the central vein and enter the fetal adrenal cortex to form the adrenal medulla, which is detectable by the eighth week. The adrenal medulla at this time is composed of sympathogonia and pheochromoblasts, which then mature into pheochromocytes. The cells appear in rosette-like structures, with the more primitive cells occupying a central position. Storage granules can be found in these cells at 12 weeks. The adrenal medullas are very small and amorphous at birth but develop into recognizable adult form by the sixth month of postnatal life.
Pheochromoblasts and pheochromocytes also collect on both sides of the aorta to form the paraganglia. These cells collect principally at the origin of the mesenteric arteries and at the aortic bifurcation where they fuse anteriorly to form the organ of Zuckerkandl, which is quite prominent during the first year of life. Pheochromocytes (chromaffin cells) also are found scattered throughout the abdominal sympathetic plexi as well as in other parts of the sympathetic nervous system.
The anatomic relationships between the adrenal medulla and the adrenal cortex differ in different species. In mammals, the medulla is surrounded by the adrenal cortex, and in humans, the adrenal medulla occupies a central position in the widest part of the gland, with only small portions extending into the narrower parts. The mass of adrenal medullary tissue in both adult adrenal glands averages about 1000 mg (about 15% of the total weight of both adrenal glands), although the proportions vary from individual to individual. There is no clear demarcation between cortex and medulla. A cuff of adrenal cortical cells usually surrounds the central vein within the adrenal medulla, and there may be islands of cortical cells elsewhere in the medulla.
The chromaffin cells or pheochromocytes of the adrenal medulla are large ovoid columnar cells arranged in nests, alveoli, or cords around a rich network of capillaries and venous sinusoids that drain blood from the adrenal cortex. Pheochromocytes have large nuclei and a well-developed Golgi apparatus. Their cytoplasm contains large numbers of vesicles (granules) that measure 100 to 300 nm in diameter and appear similar to the neurosecretory granules seen in peripheral sympathetic nerves. Catecholamines (epinephrine and/or norepinephrine) comprise about 20% of the mass of neurosecretory vesicles. Vesicles containing norepinephrine appear darker than those containing epinephrine. The vesicles also contain proteins, lipids, and adenosine triphosphate (ATP), as well as chromogranins, neuropeptide Y, enkephalins, and proopiomelanocortin (along with related peptides such as adrenocorticotropic hormone [ACTH] and β-endorphin).
The cells of the adrenal medulla are innervated by preganglionic fibers of the sympathetic nervous system, which release acetylcholine and enkephalins at the synapses. Most of these fibers arise from a plexus in the capsule of the posterior surface of the gland and enter the adrenal glands in bundles of 30 to 50 fibers without synapsing. They follow the course of the blood vessels into the medulla without branching into the adrenal cortex. Some reach the wall of the central vein, where they synapse with small autonomic ganglia. However, most fibers end in relationship to the pheochromocytes.
The adrenal gland is perfused by the superior, middle, and inferior adrenal branches of the inferior phrenic artery, directly from the aorta and from the renal arteries. On reaching the adrenal gland, these arteries branch to form a plexus under the capsule supplying the adrenal cortex. A few of these vessels, however, penetrate the cortex, passing directly to the medulla. The medulla is also nourished by branches of the arteries supplying the central vein and cuff of cortical tissue around the central vein. Capillary loops passing from the subcapsular plexus of the cortex also supply blood as they drain into the central vein. Most of the blood supply to adrenal medullary cells is via a portal vascular system that arises from the capillaries in the cortex. There is also a capillary network of lymphatics that drain into a plexus around the central vein.
Norepinephrine is converted to epinephrine by the enzyme phenylethanolamine-N-methyltransferase (PNMT). In mammals, the expression of PNMT is induced by cortisol. Chromaffin cells that produce epinephrine are exposed to higher concentrations of cortisol from capillaries draining adrenocortical cells, whereas chromaffin cells containing norepinephrine are supplied by arteries that course directly to the adrenal medulla (see sections on biosynthesis and decretion, later).
The central vein of the right adrenal is short and drains directly into the vena cava with about 5% having multiple veins. About 5% of right adrenal veins drain into the hepatic vein. For the left adrenal gland, the vein is somewhat longer and drains into the left renal vein.
Hormones of the Adrenal Medulla and Paraganglia
Catecholamines are molecules that have a catechol nucleus consisting of benzene with two hydroxyl side groups plus a side-chain amine. Catecholamines include dopamine, norepinephrine, and epinephrine (see Figure 11–2).
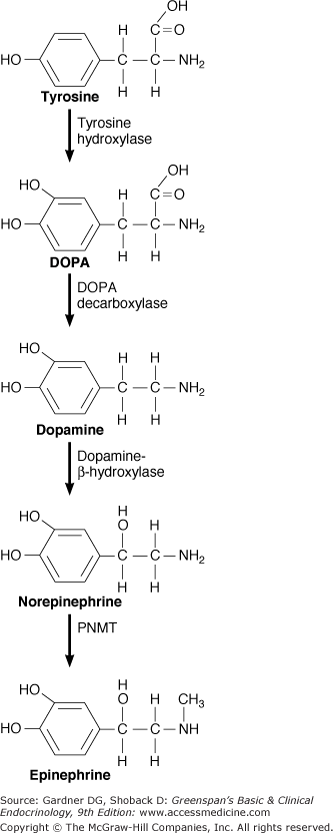
Epinephrine is the main hormone secreted by the normal adrenal medulla. The proportions of epinephrine and norepinephrine found in the adrenal medulla vary with the species; in humans, the adrenal medulla contains 15% to 20% norepinephrine.
Norepinephrine is found primarily in the central nervous system and in peripheral sympathetic paraganglia and nerves. Dopamine is found in the adrenal medulla and in sympathetic neurons as a precursor to norepinephrine. It is present in high concentrations in the brain, in specialized neurons in the sympathetic ganglia, and in the carotid body, where it serves as a neurotransmitter. Dopamine is also present in the proximal renal tubule where it promotes sodium excretion and in the gastrointestinal tract where it serves a paracrine function.
Chromogranin A (CgA) is a peptide that is stored and released with catecholamines during exocytosis; catestatin is a fragment of the CgA prohormone that inhibits further catecholamine release by acting as an antagonist at the neuronal cholinergic receptor. CgA levels tend to be somewhat higher in patients with hypertension than in matched normotensive individuals. CgA has become a valuable tumor marker, particularly for patients with paragangliomas that are otherwise nonsecretory.
The catecholamines are synthesized from tyrosine, which may be derived from ingested food or synthesized from phenylalanine in the liver. Tyrosine circulates at a concentration of 1 to 1.5 mg/dL of blood. It enters neurons and chromaffin cells by an active transport mechanism and is converted to l-dihydroxyphenylalanine (l-DOPA). The reaction is catalyzed by tyrosine hydroxylase, which is transported via axonal flow to the nerve terminal. Tyrosine hydroxylase is the rate-limiting step in catecholamine synthesis. It is transcriptionally activated by acetylcholine through the nicotinic cholinergic receptor, which in turn activates protein kinase A via cAMP. Tyrosine hydroxylase activity may be inhibited by a variety of compounds; alpha methyltyrosine (metyrosine) is an inhibitor that is sometimes used in the therapy of metastatic or unresectable pheochromocytomas.
DOPA is converted to dopamine by the enzyme aromatic l-amino acid decarboxylase (dopa decarboxylase). This enzyme is found in all tissues, with the highest concentrations in liver, kidney, brain, and vas deferens. The various enzymes have different substrate specificities depending on the tissue source. Competitive inhibitors of DOPA decarboxylase, such as methyldopa, are converted to substances (an example is α-methylnorepinephrine) that are then stored in granules in the nerve cell and released in place of norepinephrine. These products (false transmitters) were thought to mediate the antihypertensive action of drugs at peripheral sympathetic synapses but are now believed to stimulate the alpha receptors of the inhibitory corticobulbar system, reducing sympathetic discharge peripherally.
Dopamine enters granulated storage vesicles where it is hydroxylated to norepinephrine by the enzyme dopamine-β-hydroxylase (DBH), which is found within the vesicle membrane. Norepinephrine is then actively transported into the vesicle by vesicular monoamine transferase (VMAT), which is located in the lipid bilayer of the vesicle wall. Norepinephrine constantly diffuses out of the vesicle, so it is best to consider it being concentrated in the vesicle, rather than being stored there. The granulated storage vesicle migrates to the cell surface and secretes its contents via exocytosis; both norepinephrine and DBH are released during exocytosis. After secretion, most norepinephrine is avidly recycled back into the nerve. Normally, about 93% of circulating norepinephrine originates from diffusion out of nonadrenal sympathetic nerve cells and synapses, while only 7% originates from the adrenal medulla.
Norepinephrine constantly diffuses out from storage granules into the cytoplasm. In most cells of the adrenal medulla, cytoplasmic norepinephrine is converted to epinephrine, catalyzed by PNMT. Epinephrine can then return to the vesicle, diffuse from the cell, or undergo catabolism. High concentrations of cortisol enhance the expression of the gene encoding PNMT. Cortisol is present in high concentrations in most areas of the adrenal medulla due to venous blood flow from the adjacent adrenal cortex. This accounts for the fact that in the normal human adrenal medulla, about 80% of the catecholamine content is epinephrine and only 20% is norepinephrine. In mouse models, the administration of exogenous corticosteroids appears to increase adrenal medullary PNMT expression, despite suppression of ACTH and endogenous adrenal cortisol production.
Paragangliomas rarely secrete epinephrine, because they do not have high local concentrations of cortisol to induce PNMT expression. Pheochromocytomas secrete epinephrine and norepinephrine in various ratios. Interestingly, after adrenalectomy, recurrent pheochromocytoma can secrete epinephrine without any surrounding adrenal cortex, indicating that the induction of PNMT may continue in subsequent cell lineages without high local levels of cortisol. After both adrenal glands are resected, serum epinephrine levels abruptly fall to nearly zero, whereas norepinephrine concentrations do not decline significantly, since most circulating norepinephrine originates from leakage out of sympathetic nerves and synapses.
The enzyme PNMT is found in many tissues outside the adrenal medulla. PNMT that is identical to adrenal PNMT has been found in the lung, kidney, pancreas, and cancer cells. Therefore, nonadrenal tissue is capable of synthesizing epinephrine if norepinephrine is available as a substrate. However, nonadrenal production of epinephrine usually contributes minimally to circulating levels. PNMT is found in human lung, and glucocorticoids increase its expression, thereby increasing lung epinephrine. This may possibly contribute to the bronchial dilation that is induced by glucocorticoids (inhaled or systemic). PNMT activity is also found in red blood cells, where its activity is increased in hyperthyroidism and decreased in hypothyroidism. Renal PNMT activity is such that up to one-half of the epinephrine found in normal urine may be the product of renal conversion from norepinephrine.
In the adrenal medulla, catecholamine stores can be depleted during prolonged hypoglycemia. Biosynthesis of catecholamines increases during nerve stimulation by activation of tyrosine hydroxylase. Prolonged stimulation leads to the induction of increased amounts of this rate-limiting enzyme.
Catecholamines are not truly stored in intracellular vesicles. Rather, they exist in a dynamic equilibrium between the vesicle and the surrounding cytoplasm. Catecholamines continuously leak out of the vesicles into the cytoplasm but reenter and become concentrated in the vesicles by the action of two vesicular monoamine transporters: VMAT-1 and VMAT-2. Both are expressed in the adrenal medulla, but only VMAT-2 is expressed in sympathetic nerves.
In the adrenal medulla, the percentage of stored epinephrine (vs total catecholamines) varies widely among different species. In the human adrenal medulla, epinephrine comprises about 80% of stored catecholamines. Epinephrine and norepinephrine are stored in different cells. Morphologic differences in epinephrine and norepinephrine storage vesicles are visible on light microscopy after proper staining. Cells that store predominantly epinephrine tend to be adjacent to intramedullary sinusoidal vessels that bathe the cells in blood from the adrenal cortex; such blood contains cortisol that induces the enzyme PNMT that catalyzes the conversion of norepinephrine to epinephrine in the cytoplasm such that these vesicles contain predominantly epinephrine. Adrenal medullary cells that contain predominantly norepinephrine tend to be farther away from such vessels.
Norepinephrine is also found in the synaptic vesicles of postganglionic autonomic nerves in organs that have rich sympathetic innervations: the heart, salivary glands, vascular smooth muscle, liver, spleen, kidneys, and muscles. A single sympathetic nerve cell may have up to 25,000 synaptic bulges along the length of its axon; each synapse synthesizes norepinephrine and stores it in adrenergic synaptic vesicles adjacent to target cells.
Catecholamine biosynthesis is coupled to secretion, so that the stores of norepinephrine at the nerve endings remain relatively unchanged even in the presence of marked nerve activity. The adrenal medulla contains catecholamines at a concentration of about 0.5 mg/g; the spleen, vas deferens, brain, spinal cord, and heart contain 1 to 5 μg/g; and the liver, gut, and skeletal muscle contain 0.1 to 0.5 μg/g. Catecholamines are stored in electrondense granules approximately 1 μm in diameter that contain catecholamines and ATP in a 4:1 molar ratio, as well as several neuropeptides, calcium, magnesium, and water-soluble proteins called chromogranins (see earlier). The interior surface of the membrane contains DBH and ATPase. The Mg2+-dependent ATPase facilitates the uptake and inhibits the release of catecholamines by the granules. Adrenal medullary granules appear to contain and release a number of active peptides including adrenomedullin, ACTH, vasoactive intestinal peptide (VIP), chromogranins, and enkephalins. The peptides derived from chromogranins are physiologically active and may modulate catecholamine release.
(Figure 11–3 and Table 11–1)
Preganglionic nerve fibers terminate in adrenal medullary cells, where they release acetylcholine at synapses and stimulate adrenal medullary receptors. Activation of the receptors depolarizes the cell membranes and causes an influx of calcium ions. The increased intracellular concentration of calcium triggers exocytosis of the neurosecretory vesicles with consequent release of their contents: catecholamines, chromogranins, and soluble DBH. Vesicular membrane-bound DBH is not released during exocytosis.
Figure 11–3
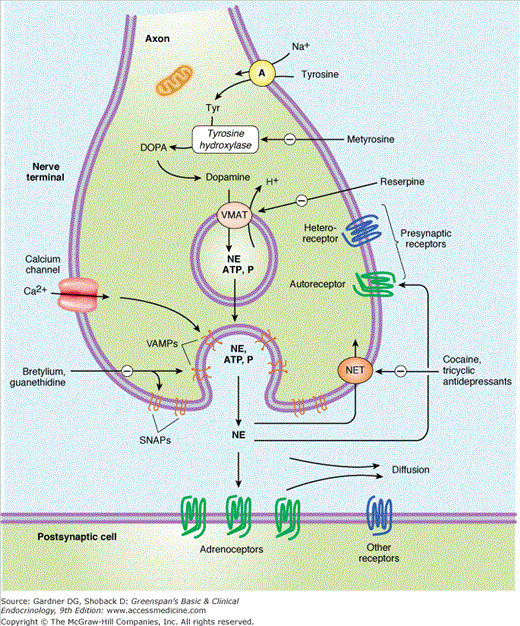
Schematic diagram of a noradrenergic junction of the peripheral sympathetic nervous system (not to scale). The neural axons form synaptic junctions with cells in target organs. The neurotransmitter is norepinephrine, which originates through de novo synthesis from tyrosine or from norepinephrine that has been secreted and reabsorbed by a norepinephrine transporter (NET). Tyrosine is transported into the noradrenergic endings or varicosities by a sodium-dependent carrier (A). In the cytoplasm, tyrosine hydroxylase converts tyrosine to DOPA that is converted by DOPA decarboxylase to dopamine. Dopamine, as well as norepinephrine (NE) is transported into the vesicles by vesicular monoamine transferase (VMAT). Dopamine is converted to NE in the vesicle by dopamine-β-hydroxylase. Exocytosis of the neurosecretory vesicle with release of transmitter occurs when an action potential opens voltage-sensitive calcium channels and increases intracellular calcium. Fusion of vesicles with the surface membrane results in expulsion of norepinephrine, cotransmitters, and dopamine-β-hydroxylase. After release, norepinephrine diffuses out of the cleft or is transported into the cytoplasm of the nerve itself (uptake 1) or transported into the postjunctional target cell (uptake 2). Regulatory receptors are present on the presynaptic terminal (SNAPs, synaptosome-associated proteins; VAMPs, vesicle-associated membrane protein). Not shown: In adrenal medullary and pheochromocytoma cells, norepinephrine constantly diffuses from the vesicle into the cytoplasm where it may be converted to epinephrine by phenylethanolamine-N-methyltransferase (PNMT). Alternatively, cytoplasmic membrane-bound catecholamine-O-methyltransferase (COMT) can metabolize epinephrine and norepinephrine directly into metanephrine and normetanephrine, respectively, which are then released.
(Reproduced, with permission, from Katzung BJ, ed. Basic & Clinical Pharmacology. 11th ed. McGraw-Hill; 2009.)
| Norepinephrine | Epinephrine | |
| Healthy | ||
| Subjects | ||
| Basal | 150-400 pg/mL (0.9-2.4 nmol/L) | 25-100 pg/mL (0.1-0.6 nmol/L) |
| Ambulatory | 200-800 pg/mL (1.2-4.8 nmol/L) | 30-100 pg/mL(0.1-1 nmol/L) |
| Exercise | 800-4000 pg/mL (4.8-24 nmol/L) | 100-1000 pg/mL (0.5-5 nmol/L) |
| Symptomatic hypoglycemia | 200-1000 pg/mL (1.2-6 nmol/L) | 1000-5000 pg/mL (5-25 nmol/L) |
| Patients | ||
| Hypertension | 200-500 pg/mL (1.2-3 nmol/L) | 20-100 pg/mL (0.1-0.6 nmol/L) |
| Surgery | 500-2000 pg/mL (3-12 nmol/L) | 199-500 pg/mL (0.5-3 nmol/L) |
| Myocardial infarction | 1000-2000 pg/mL (6-12 nmol/L) | 800-5000 pg/mL (4-25 nmol/L) |
In the adrenal medulla, exocytosis of the neurosecretory vesicles releases epinephrine and norepinephrine into the circulation. In normals, over 90% of circulating epinephrine is derived from the adrenal medulla, whereas adrenal medullary secretion accounts for only about 7% of circulating norepinephrine. Catecholamines also diffuse out of the neurosecretory vesicles into the cytoplasm. Catecholamines in the cytoplasm may reenter a vesicle via VMAT or may be metabolized by COMT into catecholamine metabolites: metanephrine and normetanephrine, which gradually diffuse out of the cell and enter the circulation.
Adrenal medullary catecholamine secretion increases with exercise, angina pectoris, myocardial infarction, hemorrhage, ether anesthesia, surgery, hypoglycemia, anoxia and asphyxia, and many other stressful stimuli. The rate of secretion of epinephrine increases more than that of norepinephrine in the presence of hypoglycemia and most other stimuli. However, during hypoxia, the adrenal medullary cells preferentially release norepinephrine. Such release is mainly not by exocytosis but rather by leakage of norepinephrine from neurosecretory vesicles into the cytoplasm and out of the cell into the circulation. The preferential release of norepinephrine may be because the adrenal medullary cells that concentrate norepinephrine are located farther from blood vessels and may be more susceptible to hypoxic injury. (See Storage of Catecholamines, earlier.)
In sympathetic neurons, exocytosis of the neurosecretory vesicle releases norepinephrine into the synapse. Norepinephrine that has been secreted into a synapse by exocytosis is either reabsorbed via norepinephrine transporters (NETs) or leaks out of the synapse into the circulation. Such synaptic leakage accounts for the majority of circulating norepinephinre in normal individuals.
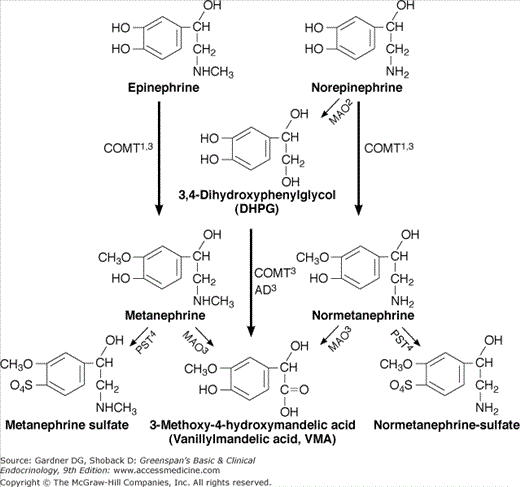
In the adrenal medulla, catecholamines are metabolized to metanephrines primarily by membrane-bound catecholamine-O-methyltransferase (COMT): epinephrine to metanephrine and norepinephrine to normetanephrine. The metanephrine metabolites subsequently leak into the circulation. Over 90% of circulating metanephrine and about 40% of circulating normetanephrine is produced by the adrenal medulla.
Catecholamines that are released at the synapse bind to their receptors with relatively low affinity and dissociate rapidly (see Figure 11–4). About 10% of norepinephrine escapes from synapses into the systemic circulation (see later). About 90% of synaptic norepinephrine is reabsorbed by the nerves from which they were released or by the target cells. Cytoplasmic norepinephrine is metabolized to 3,4-dihydroxyphenylglycol (DHPG) at the outer mitochondrial membrane by the enzyme monoamine oxidase (MAO) that regulates the norepinephrine content of neurons. DHBG leaks from the neuron and is absorbed by the liver and other tissues that convert it to VMA (see later) that is excreted in the urine. In normals, most urinary VMA is derived from DHPG that leaks from sympathetic nerves.
Sympathetic nerves in abdominal organs, particularly the intestines and pancreas, account for nearly half the body’s production of norepinephrine. Intestinal cells contain sulfatases that conjugate norepinephrine to norepinephrine-SO4 that enters the portal vein and bypasses the liver to be excreted in the urine. Intestinal cells also conjugate normetanephrine to normetanephrine-SO4.
In pheochromocytomas, catecholamines that leak out of the neurosecretory vesicles are methylated by membrane-bound COMT: epinephrine to metanephrine and norepinephrine to normetanephrine. These metanephrine metabolites continuously leak directly into the circulation, in contrast to catecholamines that are secreted intermittently. The pheochromocytoma’s continuous excretion of metanephrines accounts for the superior sensitivity of plasma or urine-fractionated free metanephrine testing in the diagnosis of pheochromocytoma. In patients with pheochromocytoma, over 90% of circulating total metanephrines are derived from the tumor itself, rather than from peripheral metabolism.
Following release into the circulation, catecholamines bind to albumin and other proteins with low affinity and high capacity. Catecholamines are quickly removed from the bloodstream and have a circulating half-life of less than 2 minutes. Although some free catecholamines are excreted directly into the urine, most are actively transported from the circulation into other cells where they are metabolized. Organic cation transporters (OCTs) are largely responsible for removing catecholamines from the circulation; they are expressed in many tissues, particularly the liver. Following active transport into a cell, catecholamines and their immediate metabolites undergo further metabolism. The main extra-adrenal/neuronal catecholamine metabolic pathway is initially via the enzyme MAO that converts both epinephrine and norepinephrine to DHPG. DHPG is further metabolized by soluble COMT to 3-methoxy-4-hydroxyphenylglycol (MHPG) that is converted by aldehyde dehydrogenase (AD) to vanillylmandelic acid (VMA) that is excreted in the urine.
Catecholamines, metanephrines, and MHPG also undergo conjugation of their phenolic hydroxyl group with sulfate or glucuronide; this reaction occurs mainly in gastrointestinal cells. To a lesser extent, catecholamines are also converted to metanephrines via a soluble form of COMT, an enzyme found in most tissues, especially blood cells, liver, kidney, and vascular smooth muscle. The soluble form of COMT is less active than the membrane-bound form of COMT found in the adrenal medulla and pheochromocytomas.
Catecholamines, their metabolites, and conjugates are excreted in the urine. Normally, the proportions of urine catecholamines and metabolites are approximately 50% metanephrines, 35% VMA, 10% conjugated catecholamines and other metabolites, and less than 5% free catecholamines.
(Tables 11–2 and 11–3; Figures 11–5 and 11–6)
| Receptor | Relative Agonist Potency | Effects |
|---|---|---|
| Alpha1 Type | Norepinephrine > epinephrine | ↑IP3, DAG common to all |
| Alpha1A | ||
| Alpha1B | ||
| Alpha1D | ||
| Alpha2 Type | Norepinephrine > epinephrine; clonidine | ↓cAMP; ↑K+ channels; ↓Ca2+ channels |
| Alpha2A | ||
| Alpha2B | Norepinephrine > epinephrine | ↓cAMP; ↓Ca2+ channels |
| Alpha2C | ↓cAMP | |
| Beta Type | ||
| Beta1 | Epinephrine = norepinephrine | ↑cAMP |
| Beta2 | Epinephrine >>> norepinephrine | ↑cAMP |
| Beta3 | Norepinephrine > epinephrine | ↑cAMP |
| Catecholamine Receptor | Tissue Location | Action Following Receptor Activation |
|---|---|---|
| Alpha1 | Vascular smooth muscle | Increases vasoconstriction (increases blood pressure) |
| Liver | Increases glycogenolysis and gluconeogenesis | |
| Eye | Increases ciliary muscle contraction (pupil dilation) | |
| Skin | Increases pilomotor smooth muscle contraction (erects hairs) | |
| Prostate | Increases contraction and ejaculation | |
| Uterus | Increases gravid uterus contraction | |
| Intestines | Increases sphincter tone and relaxes smooth muscle | |
| Spleen capsule | Contracts spleen volume, expelling blood | |
| Alpha2 | Preganglionic nerves | Decreases release of neurotransmitter |
| Vascular smooth muscle | Increases vasoconstriction (increases blood pressure) | |
| Pancreatic islet cells | Decreases release of insulin and glucagon | |
| Blood platelets | Increases platelet aggregation | |
| Adipose cells | Decreases lipolysis | |
| Brain | Decreases norepinephrine release | |
| Beta1 | Myocardium | Increases force and rate of contraction |
| Kidney (juxtaglomerular apparatus) | Increases secretion of renin | |
| Adipose cells | Increases lipolysis | |
| Most tissues | Increases calorigenesis | |
| Nerves | Increases conduction velocity | |
| Beta2 | Vascular smooth muscle | Decreases vasoconstriction (increases blood flow) |
| Bronchiolar smooth muscle | Decreases contraction (bronchial dilation) | |
| Liver | Increases glycogenolysis and gluconeogenesis | |
| Intestinal smooth muscle | Decreases intestinal motility; increases sphincter tone | |
| Pancreatic islet cells | Increases release of insulin and glucagon | |
| Adipose tissue | Increases lipolysis | |
| Muscles | Increases muscle contraction speed and glycogenolysis | |
| Liver and kidney | Increases peripheral conversion of T4 to T3 | |
| Uterus smooth muscle | Decreases nongravid uterine contraction (uterine relaxation) | |
| Beta3 | Adipose cells | Increases lipolysis |
| Intestinal smooth muscle | Increases intestinal motility | |
| Dopamine1 | Vascular smooth muscle | Decreases vasoconstriction (vasodilation); |
| Renal tubule | Enhances natriuresis | |
| Dopamine2 | Sympathetic nerves | Inhibits synaptic release of norepinephrine |
| Pituitary lactotrophes | Inhibits prolactin release | |
| Gastrointestinal tract | Paracrine functions |
Figure 11–5
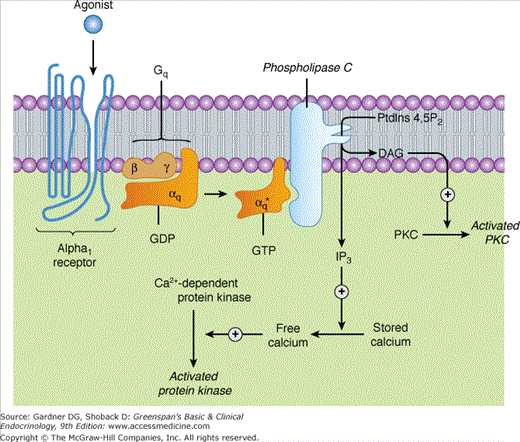
Activation of α1-adrenergic responses. Stimulation of α1 receptors by catecholamines leads to the activation of a Gq-coupling protein. The activated αq subunit of this G protein activates phospholipase C, which catalyzes the conversion of PtdIns 4,5-P2 (phosphatidylinositol 4,5-bisphosphate) to IP3 (inositol 1,4,5-trisphosphate) and DAG (diacylglycerol). IP3 stimulates the release of sequestered stores of calcium, leading to an increased concentration of cytoplasmic Ca2+ that may then activate Ca2+-dependent protein kinases that phosphorylate their substrates. DAG activates protein kinase C (PKC) (GDP, guanosine diphosphate; GTP, guanosine triphosphate). See Table 11–3 and text for physiologic effects of α1 receptor activation.
(Reproduced, with permission, from Katzung BG, ed. Basic & Clinical Pharmacology. 11th ed. McGraw-Hill; 2009.)
Figure 11–6
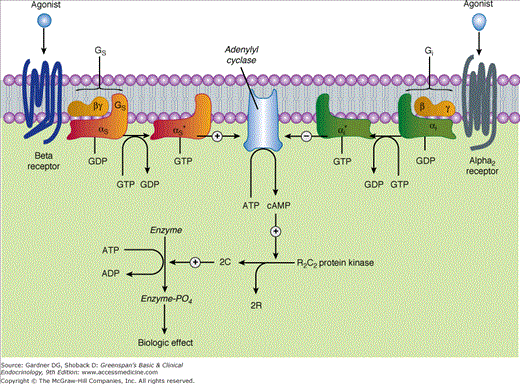
Activation of β- and α2-adrenergic responses: activation vs inhibition of adenyl cyclase. Beta receptor ligands activate the stimulatory G protein, Gs, which leads to the dissociation of its Gαs subunit, charged with GTP. The activated Gαs subunit directly activates adenyl cyclase, resulting in increased cAMP that binds to the regulatory subunit (R) of the cAMP-dependent protein kinase, leading to the liberation of active catalytic subunits (C) that phosphorylate specific enzyme substrates and modify their activity. These catalytic units also phosphorylate the cAMP response element–binding protein (CREB), which modifies gene expression. Alpha2 adrenoreceptor ligands inhibit adenyl cyclase by causing dissociation of the inhibitory G protein (Gi) into its subunits; that is, the αi subunit charged with GTP and a βγ unit. The mechanism by which these subunits inhibit adenyl cyclase is uncertain. See Table 11–3 and text for physiologic effects of β and α2 receptor activation.
(Reproduced, with permission, from Katzung BG, ed. Basic and Clinical Pharmacology. 11th ed. McGraw-Hill; 2009.)
The adrenergic receptors are G protein–coupled receptors activated by norepinephrine and epinephrine. They mediate the sympathetic fight or flight response. The finding that most cells in the body have adrenergic receptors has led to an appreciation of the important regulatory role of the peripheral sympathetic nervous system and circulating catecholamines.
Adrenergic receptors were originally classified into two groups: α and β. This classification has been expanded as subtypes of the α and β receptors have been discovered and includes α1A, α1B, α1C, α2A, α2B, α2C as well as β1 and β2, and β3. Each receptor subtype is encoded by a different gene. Adrenergic receptors are variably distributed in the central nervous system and peripheral tissues.
Norepinephrine and epinephrine are roughly equipotent in activating α and β1 receptors. Epinephrine is much more potent in activating β2 receptors and norepinephrine is more potent in activating β3 receptors (Table 11–2). The physiologic effects mediated by them are summarized in Table 11–3.
Adrenergic receptors are transmembrane proteins with an extracellular amino terminus and an intracellular carboxyl terminus. Each of their seven hydrophobic regions spans the cell membrane. Although these regions of the adrenergic receptor subtypes exhibit significant amino acid homology, differences in the fifth and sixth segments determine the specificity of agonist binding. Differences in the fifth and seventh segments determine which of the guanylyl nucleotide binding proteins (G proteins) is coupled to the receptor. G proteins consist of α, β, and γ subunits. When a hormone binds to the receptor, the β and γ subunits dissociate from the α subunits, allowing GDP to be replaced by GTP on the α subunits and causing the β and γ subunits to dissociate from it. The GTP-bound α subunits activate the postreceptor pathways (see Figure 11–5).
Alpha1 receptors are Gq-coupled receptors with three homologous subtypes: α1A, α1B, and α1C. When an agonist binds to the α1 receptor, the alpha subunit of the guanylyl nucleotide-binding protein, Gq, is released and activates phospholipase C. This enzyme catalyzes the conversion of phosphatidylinositol phosphate to 1,4,5-inositol triphosphate (IP3) and diacylglycerol. IP3 releases calcium ion from intracellular stores to stimulate physiologic responses. Diacylglycerol activates protein kinase C, which in turn phosphorylates a series of other proteins that initiate or sustain effects stimulated by the release of IP3 and calcium ion (see Figure 11–5). Prazosin and phenoxybenzamine are selective antagonists of the α1 receptor.
Alpha1 receptors are postsynaptic (target organ) receptors that mediate diverse effects, particularly arteriolar vasoconstriction. Hepatic α1 receptors stimulate glycogenolysis and gluconeogenesis. In the eye, α1 receptors stimulate contraction of the radial muscle of the iris that dilates the pupil. In the skin, α1 receptors mediate apocrine sweating and pilomotor contraction. In the genitourinary tract, α1 receptors mediate ejaculation, gravid uterine contraction, and contraction of the bladder sphincter and trigone. In the intestines, α1 receptors reduce smooth muscle contraction and increase sphincter tone, thereby promoting constipation. Alpha1 receptors are also found in the splenic capsule and mediate the contraction of the capsule (particularly during hypoxia), which contracts the splenic volume up to 20% and can expel up to 125 mL of packed red blood cells into the circulation within 3 seconds of apnea.
Sympathetic nerves secrete norepinephrine into synaptic junctions where the concentration of norepinephrine is high, thereby stimulating synaptic effector organ α1 receptors. Epinephrine has a similar effect upon α1 receptors but is secreted only by the adrenal medulla such that circulating concentrations are usually much lower than concentrations of synaptic norepinephrine. At low plasma concentrations, epinephrine predominantly stimulates β2-adrenergic receptors (causing vasodilation, see later), whereas at higher plasma concentrations, epinephrine stimulates α1 receptors sufficiently to override vasodilation and cause vasoconstriction.
Alpha2 receptors are Gi coupled receptors with three highly homologous subtypes: α2A, α2B, and α2C. Agonist binding to the α2 receptor releases Gi alpha, which inhibits the enzyme adenylyl cyclase and reduces the formation of cAMP (Figure 11–6).
Presynaptic (nerve) α2 receptors are located near the synapses of sympathetic nerves. Synaptic norepinephrine binds to this receptor, resulting in feedback inhibition of its own release. Alpha2 receptors are also found in vascular smooth muscle where they mediate vasoconstriction. Alpha2 receptors in platelets stimulate platelet aggregation and clotting. In the brain, α2 receptors are found in the locus ceruleus, cerebral cortex, and limbic system. Although their location is postsynaptic, stimulation of the α2 receptors results in reduced release of norepinephrine. Clonidine is a central α2 agonist. Yohimbine is a selective antagonist for the α2 receptor. Phentolamine is an antagonist for both α1 and α2 receptors.
In the pancreatic beta cells, activation of α2 receptors inhibits insulin secretion. This causes glucose intolerance in patients with pheochromocytoma. The gene Adra2a encodes the adrenergic α2A receptor. A polymorphism in Adra2a increases the number of α2A receptors on beta cells, which reduces insulin secretion and increases the risk of type 2 diabetes mellitus.
Beta-adrenergic receptors are postsynaptic (target organ) cell surface glycoproteins. Agonist binding to β-adrenergic receptors activates adenylyl cyclase via the Gs alpha subunit to increase the production of cAMP, which in turn converts protein kinase A to its active form. Kinase A then phosphorylates a variety of proteins, including enzymes, ion channels, and receptors (see Figure 11–6).
Beta1-adrenergic receptors are located primarily in the heart, kidneys, and adipose tissue. In the heart, β1 receptors are located in the SA and AV nodes, atrium, ventricle, and conduction system; activation of these receptors mediates an increase in heart rate and cardiac contraction with an overall increase in cardiac output. Stimulation of β1 receptors in the ventricles also increases the firing of idioventricular pacemakers such that excessive stimulation predisposes to ventricular ectopy. Triiodothyronine (T3) increases the total number of cardiac β1 receptors; this may contribute to the forceful cardiac contraction and tachyarrhythmias that are characteristic of hyperthyroidism. Beta1 receptors in the renal juxtaglomerular apparatus stimulate the release of renin that, in turn, activates the renin-angiotensin system. In fat cells, activation of β1 receptors increases lipolysis. In nerve cells, activation of β1 receptors increases nerve conduction velocity. In the brain, β1 receptors are found particularly in the nucleus accumbens and the ventral putamen (striatum).
Beta2-adrenergic receptors are particularly activated by epinephrine. They are located mainly in the smooth muscles of arterioles, veins, and bronchi where they mediate relaxation. In the heart, β2 receptors are located in the same locations as β1 receptors (see earlier) and likewise mediate increases in cardiac rate and contraction. β2 Receptors are also located in smaller coronary arteries where they mediate dilation of the coronaries, thereby increasing blood flow to cardiac muscle. Vascular β2 receptors mediate vasodilation of arterioles, particularly in skeletal muscle and the hepatic artery. Stimulation of venous β2 receptors mediates venous dilation. Stimulation of β2 receptors also causes relaxation of other smooth muscles, resulting in bronchial dilation and relaxation of the nonpregnant uterus. During exercise or hypoglycemia, epinephrine is secreted and activates hepatic β2 receptors. This stimulates glycogenolysis and gluconeogenesis for the increased glucose production that is needed for increased cardiac and skeletal muscle activity. Beta2 receptors in skeletal muscle promote increased contraction speed, glycogenolysis, and tremor. The net effect of β2 agonists (eg, epinephrine) upon liver and skeletal muscle is to increase serum glucose. Beta2-adrenergic receptors are also expressed in various areas of the brain, particularly the right hippocampus, which normally has a higher density of β2 receptors than the left hippocampus.
Beta2 receptor polymorphisms have been associated with obesity in women and differences in sensitivity to albuterol in asthmatics. Other β2 receptor polymorphisms appear to account for individual differences in the vascular response to stress and increased risk for development of hypertension-induced left ventricular hypertrophy.
Beta3-adrenergic receptors are expressed mainly in adipose tissue but also in the gallbladder, colon, central nervous system, and heart. Activation of this receptor increases energy expenditure, lipolysis, and intestinal motility. β3 receptor activation promotes lipolysis in adipose tissue and increased thermogenesis in skeletal muscle. Although β3-adrenergic receptors are found in the brains of infants, by adulthood their concentration decreases by 100-fold. Pima Indians, who are homozygous for inactivating mutations in the β3 gene, have an earlier onset of type 2 diabetes.
Five different dopamine receptors have been identified (D1 to D5), each encoded by a different gene. Dopamine receptors are found throughout the brain (including the anterior pituitary), arteries, proximal renal tubules, and gastrointestinal tract. D1 and D2 receptors are the predominant subtypes.
D1-like family of receptors (D1 and D5) couples to Gs that activates adenyl cyclase, thereby increasing intracellular cyclic AMP (cAMP). D1 receptors mediate vasodilation in the coronary, renal, cerebrovascular, and mesenteric arteries. D1 receptors in the proximal renal tubule enhance natriuresis. The D1 receptor is the most abundant dopamine receptor in the brain. Intravenous therapeutic dopamine at low doses predominantly stimulates D1 receptors, causing vasodilation. (However, high-dose therapeutic dopamine sufficiently occupies α and β receptors to cause vasoconstriction and increase blood pressure.) The A48G polymorphism of the D1 receptor gene has been associated with essential hypertension, with the 48G allele being found more frequently in hypertensive patients, presumably through a negative effect on the proximal renal tubule’s excretion of sodium.
D2-like family of receptors (D2, D3, D4) couple to a different G protein, Gi that inhibits adenyl cyclase, thereby reducing intracellular cAMP, opening potassium channels, and decreasing the influx of calcium. D2 receptors are predominately presynaptic receptors that are located on sympathetic nerves. Activation of D2 receptors inhibits the synaptic release of norepinephrine and also inhibits the transmission of nerve impulses at sympathetic ganglia. Pituitary lactotroph cells also express D2 receptors, whose activation inhibits the secretion of prolactin. Activation of other D2 receptors in the brain causes vomiting. D2 receptors are also found throughout the stomach, small intestines, and proximal colon, where the dopaminergic system is believed to work in a paracrine fashion.
The rate of catecholamine secretion largely determines sympathoadrenal activity. However, adrenergic receptors and postreceptor events are sites of fine regulation.
As noted earlier, norepinephrine and epinephrine released during nerve stimulation bind to presynaptic alpha2 receptors and reduce the amount of norepinephrine released. CgA is secreted from neurosecretory vesicles, along with catecholamines. Catestatin, a fragment of CgA, blocks postreceptor cells’ cholinergic receptors, thereby inhibiting sympathoadrenal activity.
The binding of a catecholamine agonist to its receptor reduces the number of receptors on the effector cell surface by a process called down regulation. Catecholamine receptor antagonists do not cause down regulation of receptor expression. The mechanisms involved in some of these changes are known. For example, phosphorylation of the β-adrenergic receptor by β-adrenergic receptor kinase results in sequestration of the receptors into membrane vesicles, where they are internalized and degraded. The phosphorylated receptor also has a greater affinity for β-arrestin, another regulatory protein, which prevents its interaction with Gsα.
Thyroid hormone increases the number of β receptors in the myocardium. Estrogen, which increases the number of alpha receptors in the myometrium, also increases the affinity of some vascular alpha receptors for norepinephrine.
(Tables 11–3 and 11–4)
| Norepinephrine | Epinephrine | |
|---|---|---|
| Systolic blood pressure | ↑ at 2500 pg/mL (15 nmol/L) | ↑ at 500 pg/mL (3 nmol/L) |
| Diastolic blood pressure | ↑ at 2500 pg/mL (15 nmol/L) | ↓ at 500 pg/mL (3 nmol/L) |
| Pulse | ↓ at 2500 pg/mL (15 nmol/dL) | ↑ at 250 pg/mL (1.5 nmol/L) |
| Plasma glucose | ↑ at 2500 pg/mL (15 nmol/L) | ↑ at 250-500 pg/mL (1.5-3 nmol/L) |
Norepinephrine activates α1-adrenergic receptors, increasing the influx of calcium into the target cell. Vascular α1-adrenergic receptors are found in the heart, papillary dilator muscles, and smooth muscle. Activation of α1-adrenergic receptors causes hypertension, increased cardiac contraction, and dilation of the pupils. Alpha1-adrenergic activation stimulates sweating from nonthermoregulatory apocrine stress sweat glands, which are located on the palms, axillae, and forehead. Norepinephrine also activates β1-adrenergic receptors that increase cardiac contraction and rate; stimulation of the heart rate is opposed by simultaneous vagal stimulation. Norepinephrine has less affinity for β2-adrenergic receptors. However, with higher circulating norepinephrine levels, hypermetabolism and hyperglycemia are noted. Norepinephrine also activates β3-adrenergic receptors on fat cells, causing lipolysis and an increase in serum levels of free fatty acids.
Epinephrine also stimulates α1– and β1-adrenergic receptors, with the same effects noted above for norepinephrine. However, epinephrine also activates β2 receptors, causing vasodilation in skeletal muscles. Epinephrine thus has a variable effect on blood pressure ranging from hypertension to hypotension (rare). Hypoglycemia is a strong stimulus for the adrenal medulla to secrete epinephrine, which increases hepatic glycogenolysis. Epinephrine stimulates lipolysis, resulting in increased serum levels of free fatty acids. Epinephrine also increases the basal metabolic rate. Epinephrine does not cross the blood–brain barrier well. However, high serum levels of epinephrine do stimulate the hypothalamus, resulting in unpleasant sensations ranging from nervousness to an overwhelming sense of impending doom.
Dopamine is an important central neurotransmitter and a precursor to norepinephrine and epinephrine. However, most circulating dopamine is actually derived from the gastrointestinal tract from local synthesis and dietary sources. Dopamine that is synthesized by the intestines does not function as a neurotransmitter but rather as a paracrine hormone, affecting intestinal motility, blood flow, and sodium transport. Dopamine is also synthesized in the proximal renal tubule where it is a natriuretic hormone. Dopamine synthesis is increased by a high-salt diet. The renal dopamine-paracrine system accounts for the relatively large amounts of dopamine that are normally found in the urine. Circulating dopamine is not normally a significant catecholamine. High serum concentrations of dopamine stimulate vascular D1 receptors, causing vasodilation and increased renal blood flow. Extremely high serum levels of dopamine are required to sufficiently activate vascular α1 receptors to cause vasoconstriction.
The release or injection of catecholamines generally increases heart rate and cardiac output and causes peripheral vasoconstriction, leading to an increase in blood pressure. The infusion of catecholamines also leads to a rapid reduction in plasma volume. These events are modulated by reflex mechanisms, so that, as the blood pressure increases, reflex vagal parasympathetic stimulation may slow the heart rate and tend to reduce cardiac output. Although norepinephrine usually has these effects, the effect of epinephrine may vary depending on the smooth muscle tone of the vascular system at the time. For example, in an individual with increased vascular tone, the net effect of small amounts of epinephrine may be to reduce the mean blood pressure through vasodilation, despite an increase in heart rate and cardiac output. In an individual with a reduction in vascular tone, epinephrine increases blood pressure. In addition cardiovascular function is integrated by the central nervous system, so that, under appropriate circumstances, one vascular bed may be dilated while others remain unchanged. The central organization of the sympathetic nervous system is such that its basal regulatory effects are quite discrete—in contrast to periods of stress, when stimulation may be rather generalized and accompanied by release of catecholamines into the circulation.
Catecholamines have effects on smooth muscle in tissues other than blood vessels. These effects include contraction (α1) and relaxation (β2) of uterine myometrium, relaxation of intestinal and bladder smooth muscle (β2), contraction of the smooth muscle in the bladder (α1) and intestinal sphincters, relaxation of tracheal smooth muscle (β2), and pupillary dilation (α1).
Catecholamines increase oxygen consumption and heat production. This effect is mediated by β1 receptors. Catecholamines also regulate glucose and fat mobilization from storage depots. Glycogenolysis in heart muscle and liver leads to an increase in available carbohydrate for utilization. Stimulation of adipose tissue leads to lipolysis and the release of free fatty acids and glycerol into the circulation. In humans, these effects are mediated by the β receptor. Both α1 and β2 receptors stimulate hepatic glycogenolysis and gluconeogenesis, which causes the release of glucose into the circulation. Both receptors are activated by epinephrine, which is particularly effective in raising hepatic glucose production. Plasma levels of catecholamines required to produce some cardiovascular and metabolic effects in humans are shown in Table 11–5.
Diabetes mellitus autonomic neuropathy (glucose toxicity or autoimmunity): serum norepinephrine low Recurrent hypoglycemia-associated autonomic failure: serum norepinephrine low Wolfram syndrome: diabetes insipidus, diabetes mellitus, optic atrophy, deafness (DIDMOAD): serum norepinephrine low Neurological diseases associated with autonomic neuropathy: serum norepinephrine low Pure autonomic failure: serum norepinephrine and epinephrine low Primary adrenocortical insufficiency: serum epinephrine low Congenital adrenal hyperplasia: serum epinephrine low Allgrove syndrome: achalasia, ACTH resistance, alacrima (AAA syndrome): serum epinephrine low |
Catecholamines have effects on water, sodium, potassium, calcium, and phosphate excretion in the kidney. Stimulation of the β1 receptor increases the secretion of renin from the renal juxtaglomerular apparatus, thereby activating the renin-angiotensin system. This leads to stimulation of aldosterone secretion.
Disorders of the Adrenal Medulla and Paraganglia
Epinephrine is the major catecholamine secreted by the normal adrenal medulla, and its secretion is unique to the adrenal medulla. Epinephrine is usually deficient in cortisol deficiency of any cause, because high local concentrations of cortisol in the adrenal medulla are necessary for transcription of the enzyme PNMT, which catalyzes the conversion of norepinephrine to epinephrine in the catecholamine biosynthetic pathway. Primary autoimmune adrenal cortical insufficiency produces epinephrine deficiency. Hypothalamic/pituitary ACTH deficiency also causes cortisol deficiency and consequently epinephrine deficiency.
(Table 11–5 and Figure 11–6)
Autonomic insufficiency is also know as dysautonomia. It refers to a general reduction in autonomic activity. Since the adrenal medulla is part of the autonomic nervous system, there is usually an associated deficiency in circulating epinephrine. Autonomic insufficiency most commonly occurs in patients with long-standing type 1 diabetes mellitus. Diabetic autonomic neuropathy may be due to glucose neurotoxicity, but may also be due to autoimmunity. In one study of middle-aged patients with type 1 diabetes mellitus, 52% had complement-fixing antibodies against the sympathetic ganglia and 35% had antibodies against the adrenal medulla. They have minor defects in recovery from insulin-induced hypoglycemia (Figure 11–7), but in patients with diabetes mellitus in whom the glucagon response is also deficient, the additional loss of adrenal medullary response leaves them more susceptible to bouts of severe hypoglycemia. This is the result of a decrease in the warning symptoms as well as an impaired response to the hypoglycemia (see Chapters 17 and 18). Patients with generalized autonomic insufficiency usually have orthostatic hypotension. Familial dysautonomia can manifest as orthostatic tachycardia; norepinephrine transporter deficiency has been determined to be one cause of this condition. The causes of disorders associated with autonomic insufficiency are listed in Table 11–5.
Myocardial infarction Cerebrovascular accident Arrhythmias Irreversible shock Renal failure Dissecting aortic aneurysm Acute respiratory distress syndrome |
Figure 11–7
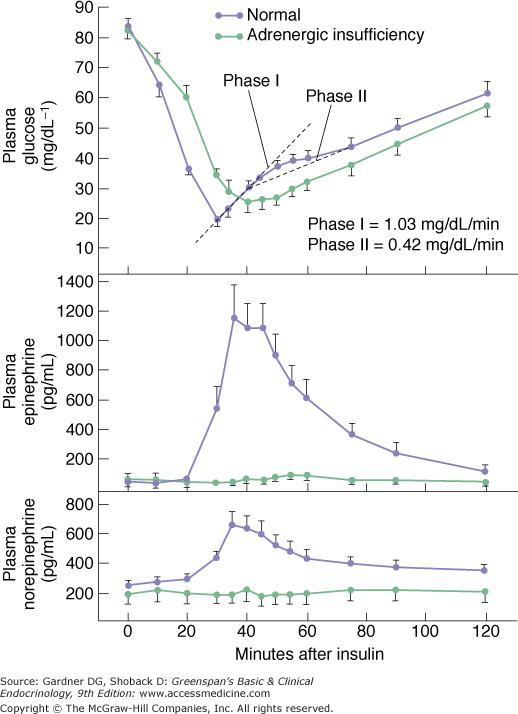
Plasma glucose, epinephrine, and norepinephrine levels after insulin administration in 14 normal subjects ( ) and 7 patients with idiopathic orthostatic hypotension (
) and 7 patients with idiopathic orthostatic hypotension ( ) with low or absent epinephrine responses. Results are expressed as mean ± SEM.
) with low or absent epinephrine responses. Results are expressed as mean ± SEM.
(Reproduced, with permission, from Polinsky RJ, et al. The adrenal medullary response to hypoglycemia in patients with orthostatic hypotension. J Clin Endocrinol Metab. 1980;51:1404.)
When a normal individual stands, a series of physiologic adjustments occur that maintain blood pressure and ensure adequate circulation to the brain. The initial lowering of the blood pressure stimulates the baroreceptors, which then activate central reflex mechanisms that cause arterial and venous constriction, increase cardiac output, and activate the release of renin and vasopressin. Interruption of afferent, central, or efferent components of this autonomic reflex results in autonomic insufficiency.
Patients with generalized autonomic insufficiency usually have orthostatic hypotension; the reduction in blood pressure on standing that causes visual disturbances, lightheadedness, and even syncope. Symptoms tend to be worse upon awakening, after meals, in hot weather, and at high altitude. Many patients have supine hypertension, even when not taking hypertensive medication. Affected patients often have a fixed heart rate, sinus tachycardia, diminished sweating, reduced metabolic rate, heat intolerance, nocturia, and erectile dysfunction. Angina pectoris can sometimes occur in the absence of coronary atherosclerosis. Patients frequently have a neurogenic bladder with associated symptoms of frequency, nocturia, hesitancy, reduced urine stream, and recurrent urinary tract infections. Patients may also have gastroparesis and disordered gastrointestinal motility. Sleep apnea is common and death can occur during sleep from reduced respiratory drive.
The diagnosis of autonomic insufficiency can usually be made clinically in patients with typical symptoms and orthostatic blood pressure changes on examination. Additionally, plasma and urine norepinephrine levels are usually very low, sometimes only 10% of average normal in severely affected individuals. Plasma epinephrine levels are often low as well.
The treatment of symptomatic orthostatic hypotension is dependent on maintenance of an adequate blood volume. Physical measures should be employed, such as raising the foot of the bed at night, crossing the legs when sitting, and wearing full-leg fitted support stockings. Adequate hydration is important. Volume expansion with fludrocortisone is the most effective drug treatment. However, fludrocortisone can cause hypokalemia, supine hypertension, and edema. Midodrine is an α1-receptor agonist that must be administered every 8 hours, due to its short half-life of 3 to 4 hours. Midodrine’s use is limited by common side effects that include severe supine hypertension, bradycardia, and erythema multiforme. Therefore, midodrine should only be used for patients with very symptomatic orthostatic hypotension who have failed other therapies. Octreotide has been used, either alone or in combination with midodrine, for patients with severe autonomic failure where other treatment measures have failed.
Pheochromocytoma and Paraganglioma
Both pheochromocytoma and non–head-neck paraganglioma are tumors of the sympathetic nervous system. Although they are similar tumors, they warrant distinction from each other to emphasize their differences in the following: (1) locations, (2) manifestations, (3) secretion profiles, (4) genetic syndromes, (5) difficulty of surgical resection, and (6) propensity to metastasize.
Pheochromocytomas are tumors that arise from the adrenal medulla, whereas non–head-neck paragangliomas arise from the ganglia of the sympathetic nervous system. Pheochromocytomas are more common (85%), and may be bilateral (17%). Patients with adrenal pheochromocytomas are less likely to develop detectable metastases (11%) compared to paragangliomas (30%).
The yearly incidence of diagnosed pheochromocytoma/paraganglioma tumors is about 2 to 8 per million; nearly half of these present as an unexplained death. The National Cancer Registry in Sweden has reported that pheochromocytomas/paragangliomas are discovered in about 2 patients per million people yearly. However, autopsy series suggest a higher incidence. The reported incidence in autopsy series has varied from about 250 cases per million to 1300 cases per million in a Mayo Clinic autopsy series. Retrospectively, 61% of pheochromocytoma/paraganglioma tumors at autopsy occurred in patients who were known to have had hypertension; about 91% had the nonspecific symptoms associated with secretory pheochromocytoma/paraganglioma tumors. In one autopsy study, a large number of patients with these tumors had nonclassic symptoms such as abdominal pain, vomiting, dyspnea, heart failure, hypotension, or sudden death (Table 11–6). Considering these autopsy data, it is clear that the majority of pheochromocytoma/paraganglioma tumors are not diagnosed during life. Tumors occur in both sexes and at any age but are most commonly diagnosed in the fourth and fifth decades.
Pheochromocytomas are chromaffin tumors that arise from the adrenal medulla. They account for 90% of all pheochromocytoma/paraganglioma tumors in adults and 70% in children. Adrenal pheochromocytomas are usually unilateral (90%). Unilateral pheochromocytomas occur more frequently in the right (65%) versus the left adrenal (35%). Right-sided pheochromocytomas have been described as producing paroxysmal hypertension more often than sustained hypertension, whereas the opposite was true for tumors arising from the left adrenal. Adrenal pheochromocytomas are bilateral in about 10% of adults and 35% of children. Bilateral pheochromocytomas are particularly common (24% overall) in patients with familial pheochromocytoma syndromes caused by certain germline mutations (see later).
Basically, the adrenal medulla is a sympathetic ganglia whose neurons secrete neurotransmitters (epinephrine and norepinephrine) into capillaries instead of synapses. The adrenal medulla is surrounded by the adrenal cortex, with which it shares a portal system that bathes it in high concentrations of cortisol. Cortisol stimulates expression of the gene encoding PNMT, the enzyme that catalyzes the conversion of norepinephrine to epinephrine. Adrenal pheochromocytomas nearly always secrete catecholamines with variable amounts of epinephrine as well as norepinephrine and their metanephrine metabolites. Metastases from adrenal pheochromocytomas usually secrete only norepinephrine and its metabolite normetanephrine; they may rarely secrete epinephrine.
Pheochromocytomas are typically encapsulated by a true capsule or a pseudocapsule, the latter being the adrenal capsule. Pheochromocytomas are firm in texture. Hemorrhages that occur within a pheochromocytoma can give the tumor a mottled or dark red appearance. A few black pheochromocytomas have been described, with the dark pigmentation believed to be due to an accumulation of neuromelanin, a catecholamine metabolite. Larger tumors frequently contain large areas of hemorrhagic necrosis that have undergone cystic degeneration; viable tumor may be found in the cyst wall. Calcifications are often present. Pheochromocytomas can invade adjacent organs, and tumors may extend into the adrenal vein and the vena cava, resulting in pulmonary tumor emboli. Pheochromocytomas vary tremendously in size, ranging from microscopic to 3600 g. The average pheochromocytoma weighs about 100 g and is 4.5 cm in diameter.
Non–head-neck paragangliomas arise from sympathetic ganglia (Figure 11–8). They account for about 10% of all pheochromocytoma/paraganglioma tumors in adults and about 30% in children. About 75% are intra-abdominal where they are often mistaken for adrenal pheochromocytomas. Paragangliomas are sometimes nonsecretory and may be confused with other neuroendocrine tumors. They arise most commonly in the perinephric, periaortic, and bladder regions but often found in the chest (25%), arising in the anterior or posterior mediastinum or the heart. Pelvic paragangliomas may involve the bladder wall, obstruct the ureters, and metastasize to regional lymph nodes. About 36% to 60% of paragangliomas are functional, secreting norepinephrine and normetanephrine. Functional status is not known to affect survival. Nonfunctional paragangliomas can often concentrate metaiodobenzylguanidine (MIBG) or secrete CgA. Paragangliomas of the bladder can cause symptoms associated with micturition. Large perinephric tumors can cause renal artery stenosis and elevations in plasma renin activity. Vaginal tumors may cause dysfunctional vaginal bleeding. Paragangliomas can rarely arise in central nervous system locations, including the sella turcica, petrous ridge, and pineal region. Cauda equina paragangliomas can cause increased intracranial pressure.
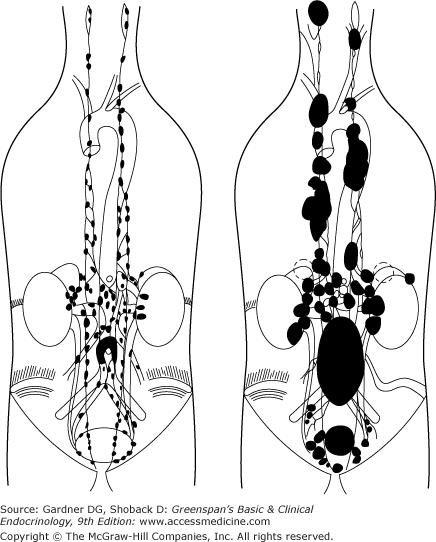
Paragangliomas commonly metastasize (30%-50%) and present with pain or a mass. They tend to metastasize to the liver, lungs, lymph nodes, and bone. PGLs can be locally invasive and may destroy adjacent vertebrae and cause spinal cord and nerve root compression. PGL metastases usually secrete norepinephrine and normetanephrine. They do not secrete epinephrine and therefore do not typically cause significant weight loss, hyperglycemia, anxiety, or tremor.
Nonchromaffin parasympathetic paragangliomas are typically found in the head and neck. Unlike chromaffin sympathetic paragangliomas, only about 5% of these tumors secrete catecholamines. Carotid body tumors are paragangliomas that arise near the carotid bifurcation. They usually present as a painless neck mass, but can injure the vagus or hypoglossal nerves. Glomus tympanicum paragangliomas arise in the middle ear and cause tinnitus and hearing loss. The tumor may be seen as a reddish mass behind an intact tympanic membrane (red drum) that blanches when pressure is applied with a pneumatic ear speculum (Brown sign). Glomus jugulare paragangliomas arise in the jugular foramen. They often cause tinnitus and hearing loss as well as compression of cranial nerves, resulting is dysphagia. Vagal paragangliomas are rare, usually presenting as a painless neck mass often associated with hoarseness or dysphagia. Head-neck paragangliomas can also arise in the larynx, nasal cavity, nasal sinuses, and thyroid gland. Head-neck PGLs themselves are of more interest to otolaryngologists than endocrinologists. But they may be seen along with sympathetic paragangliomas in familial paraganglioma syndromes, particularly those with germline succinate dehydrogenase subunit D (SDHD) mutations (see later).
Nonchromaffin paragangliomas behave differently from sympathetic paragangliomas and pheochromocytomas, although they are embryologically related to chromaffin sympathetic paragangliomas and may arise concurrently with sympathetic paragangliomas in familial paraganglioma syndromes. These tumors are less likely to be malignant, although their indolence can be deceiving; recurrence and metastases may not appear for many years. Metastases to local nodes, lungs, and bone can occur. Long-term surveillance for recurrence or metastases is recommended for all patients. These tumors can produce CgA, and preoperative determination of CgA is recommended to determine if it will be a good serum marker for tumor recurrence. Patients with cervical paragangliomas often have an underlying germline mutation in the gene encoding succinate dehydrogenase (SDH); SDHD, SDHC, and SDHB gene sequencing is recommended for all these patients (see later). Individuals harboring a germline mutation must be screened for the existence of other paragangliomas and pheochromocytomas (see “Genetic Conditions Associated with Pheochromocytomas and Paragangliomas,” later).
Neuroblastomas, ganglioneuroblastomas, and ganglioneuromas are sympathetic nervous system tumors that are related to pheochromocytomas and likewise arise from embryonal sympathogonia (see Figure 11–1). Neuroblastoma is the most common malignant disease of infancy and the third most common pediatric cancer, accounting for 15% of childhood cancer deaths. Neuroblastomas are embryonal cancers of the postganglionic sympathetic nervous system and develop from sympathetic tissue in the adrenal gland, neck, posterior mediastinum, retroperitoneum, or pelvis. These tumors differ in their degree of maturation and malignancy. Neuroblastoma tumors derive from immature neuroblasts, are generally aggressive, tend to occur in very young children, and display clinical heterogeneity. The prognosis for neuroblastoma depends on age at diagnosis, with infants under age 1 year having a high likelihood of cure with surgical excision. Similarly, children with a localized neuroblastoma may be cured surgically. Some tumors can regress spontaneously or differentiate into benign ganglioneuromas. However, older children with widespread hematogenous metastases have a very poor prognosis, as do those whose neuroblastoma harbors an amplification of the Myc proto-oncogene. Some neuroblastomas are more indolent, depending on the nature of their oncogene mutations.
Ganglioneuroblastomas are composed of a mixture of neuroblasts and more mature gangliocytes; these develop in older children and tend to run a more benign course. Ganglioneuromas are the most benign of these tumors and are composed of gangliocytes and mature stromal cells.
Despite catecholamine secretion, children with neuroblastomas tend to be more symptomatic from their primary tumor or metastases than from catecholamine secretion. Tumors tend to concentrate radiolabeled MIBG, making it a useful imaging and therapeutic agent. Treatment of malignant tumors consists of surgery, chemotherapy, external beam radiation to skeletal metastases, and high-dose 131I-MIBG therapy for patients with MIBG-avid tumors.
Hypertension, defined as either a systolic or diastolic blood pressure over 140/90 mm Hg, is an extremely common condition, affecting about 20% of all American adults and over 50% of adults over 60 years of age. The incidence of pheochromocytoma is estimated to be less than 0.1% of the entire hypertensive population, but it is higher in certain subgroups whose hypertension is labile or severe. Screening for pheochromocytomas should be considered for such patients with severe hypertension and also for hypertensive patients with suspicious symptoms (eg, headaches, palpitations, sweating episodes, or unexplained bouts of abdominal or chest pains; Table 11–7).
Young hypertensives Hypertensive patients with: Symptoms listed in Table 11–13 Weight loss Seizures Orthostatic hypotension Unexplained shock Family history of pheochromocytoma or medullary carcinoma of thyroid Neurofibromatosis and other neurocutaneous syndromes Mucosal neuromas Hyperglycemia Cardiomyopathy Marked lability of blood pressure Family history of pheochromocytoma Shock or severe pressor responses with: Induction of anesthesia Parturition Surgery Invasive procedures Radiologic evidence of adrenal mass Radiologic evidence of mass in area of paraganglia |
(Tables 11–8, 11–9, 11–10, and 11–11)
Twice yearly: physical examination with blood pressure and neck examination; plasma free metanephrine; serum calcium and albumin (serum PTH if RET codon 634 mutation); serum calcitonin (patients with intact thyroid or with medullary thyroid carcinoma) Prophylactic thyroidectomy: by age 5 years for MEN 2A and by 6 months for codon 918 mutations (MEN 2B) Yearly thyroid ultrasound for patients with an intact thyroid For rising or elevated serum calcitonin levels after thyroidectomy: close surveillance for medullary thyroid carcinoma is required. 18FDG-PET-CT fusion scans are particularly helpful For suspicious symptoms and before major surgical procedures and pregnancy: biochemical screening for pheochromocytoma with plasma free metanephrine For abnormal biochemical screening for pheochyromocytoma: MRI or CT scan of the abdomen (nonionic contrast) with thin-section adrenal cuts; 123I-MIBG SPECT or 18F-FDA PET scan to confirm the identity of small masses |
Yearly: retinal examination; plasma free normetanephrine levels yearly from age 10 to 15 years Twice yearly: physical examination with blood pressure beginning at age 5 years; plasma free normetanephrine levels beginning at age 15 years Every 2 years: MRI scanning (with intravenous contrast) of the entire brain and spinal cord (VHL types 2A and 2B VHL); MRI scanning of the abdomen For suspicious symptoms and before major surgical procedures and pregnancy: biochemical screening for pheochromocytoma with plasma free metanephrine For abnormal biochemical screening: MRI or CT scan of the abdomen (nonionic contrast) with thin-section adrenal cuts; 123I-MIBG SPECT or 18F-FDA PET scan to confirm the identity of small masses |
Twice yearly: physical and neurologic examination and blood pressure, and a careful examination of the skin for development or growth of neurofibromas Yearly: CBC with WBC differential; plasma-fractionated free metanephrines; complete eye examination with visual fields Before major surgical procedures and pregnancy: plasma-fractionated free metanephrines to screen for pheochromocytoma For abnormal biochemical screening: MRI or CT scan of the abdomen (nonionic contrast); 123I-MIBG SPECT or 18F-FDA PET scan |
Twice yearly: physical examination and blood pressure; plasma-fractionated free metanephrines and serum chromogranin A Yearly: scanning with ultrasound of the neck and abdomen on odd years and MRI or CT (nonionic contrast) of the neck, chest, abdomen, and pelvis on even years For suspicious symptoms and before major surgical procedures and pregnancy: complete biochemical screening for pheochromocytoma with fractionated plasma metanephrines and serum chromogranin A For abnormal biochemical screening: CT scan (nonionic contrast) of the chest, abdomen, and pelvis or 18F-FDG-PET with CT fusion scan; to confirm ambiguous results: 123I-MIBG SPECT or 18F-FDA PET scan |
Most pheochromocytomas occur sporadically, although a substantial proportion of these tumors are found to have developed a somatic mutation similar to those germline mutations that give rise to familial syndromes. Based on family history, it was formerly thought that only about 10% of patients with these tumors developed it as part of a genetic syndrome. With genetic testing, it is now clear that about 26% of patients with pheochromocytomas or paragangliomas carry a germline mutation associated with a familial syndrome. About 19% of patients with an apparently sporadic single pheochromocytoma and no known family history of pheochromocytoma harbor a germline mutation. For example, about 9% of patients with apparently sporadic, unilateral pheochromocytoma have been found to have von Hippel-Lindau (VHL) mutations. Genetic testing is advisable for all patients with a pheochromocytoma or paraganglioma. Genetic testing is strongly recommended for patients with extra-adrenal paragangliomas or multifocal tumors; onset of symptoms before age 45 years; prior history of head-neck paraganglioma; family history of paraganglioma, pheochromocytoma, or other tumors associated with MEN 2 or VHL. Genetic screening is also performed for patients with other manifestations of genetic syndromes noted later. Genetic screening can be done for MEN 2 RET proto-oncogene mutations, VHL mutations, and SDH (SDHB, SDHD, and SDHC) gene mutations. Neurofibromatosis is usually diagnosed clinically without genetic testing, since the NF-1 gene is very large.
The first description of a pheochromocytoma was in a patient with MEN 2. In 1882, Felix Fränkel described pheochromocytomas in an 18-year-old woman with bilateral adrenal tumors; genetic testing of four living relatives has revealed a germline RET mutation.
The prevalence of MEN 2 has been estimated at 1 in 30,000. MEN 2 is an autosomal dominant disorder that causes a predisposition to medullary thyroid carcinoma, pheochromocytoma, and other abnormalities described later. MEN 2 is caused by an activating mutation in the RET proto-oncogene on chromosome 10q11.2. RET consists of 21 exons that encode a transmembrane receptor tyrosine kinase that is expressed in neural crest tissues. The constitutive activation of RET causes hyperplasia in affected tissues, including the adrenal medulla. Additional mutation(s) are believed to be required for a pheochromocytoma to develop. The somatic loss of a tumor suppressor gene on chromosome 1p appears to be necessary for pheochromocytoma formation. Additionally, reduced expression of NF-1 has been reported in a minority of pheochromocytomas arising in patients with MEN 2 mutations.
MEN 2 kindreds can be grouped into two distinct subtypes: MEN 2A (90%) and MEN 2B (10%). Patients with MEN 2A have various single amino acid missense mutations affecting the extracellular RET domain that cause RET homodimerization and constitutive activation of its tyrosine kinase. Patients with MEN 2B have a particular missense mutation (codon 918, exon 16) affecting the intracellular domain at the RET tyrosine kinase’s catalytic site, causing constitutive activation. In either subtype, pheochromocytomas usually develop in the adrenals (96%); extra-adrenal paragangliomas are rare (4%). About 42% of patients with pheochromocytoma in MEN 2 have hypertension, usually paroxysmal. Overall, only about 53% exhibit symptoms of pheochromocytoma. Each specific type of mutation in the RET codon determines each kindred’s idiosyncrasies, such as the age at onset and the aggressiveness of medullary thyroid carcinoma.
A limited number of RET gene exons have been found to harbor mutations that are capable of causing constitutive activation of the tyrosine kinase. RET exons 10, 11, 13, 14, 15, and 16 are usually involved. Therefore, routine genetic screening searches for mutations only in these exons. If no mutation is found in these exons, the remaining 15 exons can be sequenced in a research laboratory. When an affected kindred’s RET mutation is already known, an individual in the kindred is screened for that specific mutation.
Pheochromocytomas arise in the adrenals, and extra-adrenal paragangliomas are rare in MEN 2. In patients with MEN 2, only about 4% of pheochromocytomas are found to be metastatic, possibly because of earlier detection. When pheochromocytomas develop in patients with MEN 2, they are bilateral in about two-thirds of cases; however, with close surveillance of affected kindreds, pheochromocytomas are discovered earlier and are more likely to be unilateral. After a unilateral adrenalectomy for pheochromocytoma in a patient with MEN 2, a contralateral pheochromocytoma develops in about 50% of patients an average of 12 years after the first adrenalectomy. In patients with MEN 2, adrenal pheochromocytomas produce norepinephrine and epinephrine (with its metabolite metanephrine). When screening for small pheochromocytomas, plasma catecholamine concentrations may be normal but plasma metanephrine concentrations are usually elevated, making plasma free metanephrines the screening test of choice for these patients. However, some pheochromocytomas are detected only by a 24-hour urine determination for fractionated metanephrines, catecholamines, and creatinine.
MEN 2A (Sipple syndrome)—Patients with this genetic condition develop medullary thyroid carcinoma (95%-100%), hyperparathyroidism due to multiglandular hyperplasia (35%), and pheochromocytoma (50%; range 6%-100% depending on the kindred) or adrenal medullary hyperplasia. Patients with MEN 2A also have an increased incidence of cutaneous lichen amyloidosis and Hirschsprung disease. Pheochromocytomas tend to present in middle age, often without hypertension. The incidence of hyperparathyroidism has also varied, averaging 35%.
Individuals belonging to an MEN 2A kindred should have genetic testing for RET proto-oncogene mutations by 5 years of age to determine if they carry the genetic mutation that will require prophylactic thyroidectomy and close surveillance for pheochromocytoma and hyperparathyroidism.
More than 85% of the mutations in MEN 2A families affect codon 634 in exon 11 of the RET proto-oncogene. Individuals with mutations in RET codon 630 also have a high incidence of pheochromocytoma. Pheochromocytomas also occur in most other kindreds with MEN 2A. Screening for pheochromocytoma should commence with the routine blood pressure measurements that are performed during examinations in childhood for medullary thyroid carcinoma and follow-up of hypothyroidism following thyroidectomy. At about age 15 years, affected individuals should commence yearly screening for pheochromocytoma with plasma free metanephrine determinations.
Certain RET mutations (codons 609, 768, val804met, and 891) rarely produce pheochromocytomas. Less intense screening for pheochromocytomas is required in patients with these mutations.
MEN 2B—Over 90% of patients with MEN 2B have a single amino acid substitution of methionine to threonine on codon 918 in exon 16 that affects the intracellular domain of tyrosine kinase. About 50% of such mutations are familial while the rest arise de novo and are sporadic. Patients with this genetic condition are prone to develop mucosal neuromas, pheochromocytoma, adrenal medullary hyperplasia, and aggressive medullary thyroid carcinoma. About 50% of affected patients develop all three manifestations of the syndrome. Mucosal neuromas tend to develop first and occur in most patients. They appear as small bumps on the tongue, lips and buccal mucosa, eyelids, cornea, and conjunctivae. The lips and eyelids may become diffusely thickened. Intestinal ganglioneuromatosis occurs and alters intestinal motility, causing diarrhea or constipation and occasionally megacolon. Affected individuals have a marfanoid habitus with associated spinal scoliosis or kyphosis, pectus excavatum, a high-arched foot and talipes equinovarus (club foot) deformities. In patients with MEN 2B, medullary thyroid carcinoma tends to be aggressive and occurs at an earlier age than in patients with MEN 2A. Members of kindred with MEN 2B should immediately have genetic testing for their family’s RET proto-oncogene mutation. If an individual is found to carry the family’s RET mutation, early prophylactic thyroidectomy is advisable. In kindreds with MEN 2B, infants are screened for the mutation at birth. For affected infants, prophylactic thyroidectomy is performed by 6 months of age.
All individuals carrying a RET proto-oncogene mutation must have close medical surveillance (see Table 11–8). See Chapter 22: Multiple Endocrine Neoplasia.
VHL disease is inherited as an autosomal dominant trait that predisposes its carriers to develop tumors in multiple tissues. Although certain VHL kindreds are predisposed to develop pheochromocytomas, it is not the dominant tumor of this syndrome. Pheochromocytomas develop only in patients with type 2 VHL (see later), and these are different from sporadic pheochromocytomas in that they are less likely to be malignant, more likely to be bilateral, and more likely to present at an earlier age. Although most VHL-associated pheochromocytomas arise in the adrenal, extra-adrenal paragangliomas sometimes arise; these usually occur along the sympathetic chain, but head-neck parasympathetic paragangliomas have been described. The mean age of presentation for a pheochromocytoma in type 2 VHL is 28 years; the youngest reported patient was 5 years old.
The different types and subtypes of VHL are as follows:
Type 1 VHL—Affected members of families with type 1 VHL do not develop pheochromocytomas. They tend to have loss-of-function VHL gene mutations, particularly gene deletions, frameshifts, or truncations.
Type 2 VHL—Affected members of families with type 2 VHL mutations are prone to develop pheochromocytomas. These patients carry VHL missense mutations. Type 2 VHL may be divided into subtypes as follows:
Type 2A VHL—Pheochromocytomas, hemangioblastomas, low risk of renal cell carcinoma.
Type 2B VHL—Pheochromocytomas, hemangioblastomas, high risk of renal cell carcinoma.
Type 2C VHL—Pheochromocytomas, no hemangioblastomas, no renal cell carcinomas.
The prevalence of VHL is about 1 in 30,000 persons. VHL disease carriers are predisposed to multicentric hemangioblastomas in the retina (retinal angiomas), cerebellum, and spinal cord. Renal cysts and renal clear-cell carcinoma commonly develop.
Multiple cysts and nonfunctional neuroendocrine tumors may develop in the pancreas. Endolymphatic sac tumors may occur, resulting in hearing loss, vertigo, or ataxia. Adnexal cystadenomas of probable mesonephric origin (APMO) are found in many women with VHL. APMOs may develop in the ovary, broad ligament, vagina, uterine cervix, and vulva. Equivalent epididymal cystadenomas occur in men with VHL.
Genetic testing for VHL disease should be done in children born to a parent with known VHL disease. If VHL disease is suspected, but the family’s VHL mutation is not known, the patient must be screened for point mutations by direct VHL gene sequencing of the entire coding region and splice junctions.
The clinical diagnosis of VHL disease is made when a patient with a known VHL
Related posts:
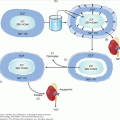

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


