Fig. 14.1
A patient with benign bilateral adrenal lesions. He has a symptomatic left adrenal myelolipoma and underwent a left adrenalectomy. The 5 cm asymptomatic lesion on the right side was left alone. (a & b): CT scan – a large mass occupying most of the left abdomen, pushing the left kidney inferiorly and the gut toward the right side of the abdomen. (c): Intraoperative photo of a large adrenal lesion that is not amenable to a minimally invasive surgical resection. (d): Gross specimen (e): Histology slide – adrenal myelolipoma: mature adipocytes are admixed with hematopoietic cells
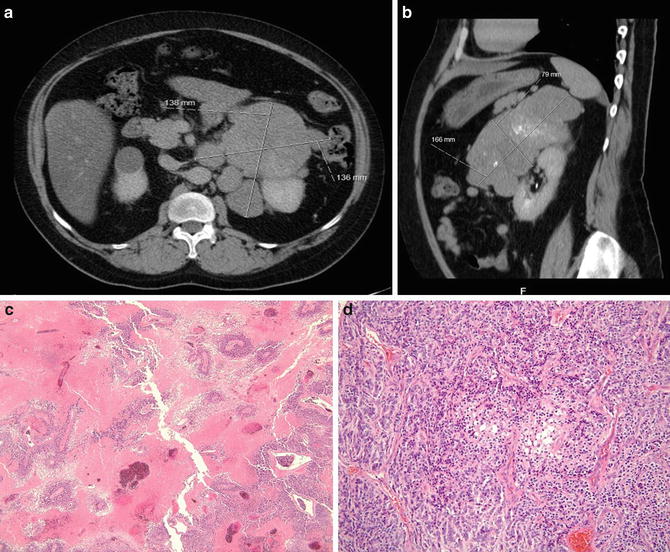
Fig. 14.2
A 33-year-old patient presented with Cushing’s disease and was found to have a 17 cm left adrenal mass (adrenocortical carcinoma – ACC). The patient underwent a hand-assisted laparoscopic left adrenalectomy and nephrectomy with clear margins. (a & b): CAT scan shows large lobulated left adrenal mass (ACC) (c): Histology – low-power photomicrograph (40×) of adrenal cortical carcinoma areas of extensive confluent necrosis (d): Histology – medium-power photomicrograph (200X) of an adrenal cortical carcinoma demonstrating cellular pleomorphism and atypia, mitotic activity, and trabecular growth pattern. The tumor also exhibited capsular invasion, and tumor cells showed a typical adrenocortical immunohistochemical profile with positivity for inhibin, melan-A, and synaptophysin, and negativity for EMA and pancytokeratin
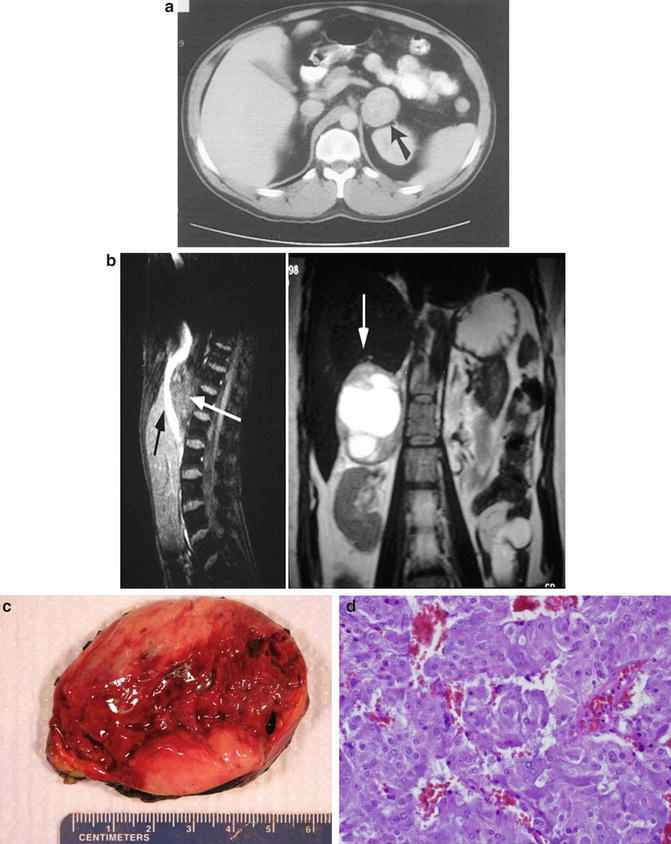
Fig. 14.3
Patient with a left pheochromocytoma (a): CT scan – black arrow is pointing toward the left adrenal mass. (b): MRI – white arrow pointing toward the left adrenal mass. (c): Gross photo (d): Histology – this photomicrograph of a pheochromocytoma illustrates the typical “Zellballen” architectural pattern in which balls of tumor cells are supported by a rich vascular framework. The cytoplasm has a finely granular appearance, and nuclei demonstrate a stippled chromatin pattern. Though mitotic activity is typically sparse in these tumors, note the enlarged and hyperchromatic nuclei (hematoxylin and eosin stain, original magnification 200×)
Historically, adrenal scintigraphy with radiocholesterol was used to differentiate benign from malignant adrenal masses. However, a lack of widespread expertise, lack of tracer availability, poor resolution, prolonged time needed to complete the procedure (required over a period of 1 week to complete), and high radiation dose needed are the main limitations of this imaging modality [3, 4]. However, when pheochromocytoma is suspected, scintigraphy with I-123 or I-131 metaiodobenzylguanidine (MIBG) should be performed [3] (Fig. 14.4). Lastly, 18F-fluorodeoxyglucose positron emission tomography (FDG-PET) scan might be very helpful in differentiating malignant (adrenal carcinoma and metastasis) from benign lesions in patients with radiological undetermined adrenal lesion [3, 4] (Fig. 14.5). Adrenal FDG uptake is considered to be malignant when the intensity is higher than hepatic uptake. False positives may be seen in patients with sarcoidosis, tuberculosis, pheochromocytoma, and some adenomas, while false negatives may be seen in patients with necrotic and hemorrhagic cancer lesions as well as in some primary malignant lesions. In a multicenter prospective study of 77 patients who underwent adrenalectomy, preoperative 18F-FDG-PET imaging successfully distinguishes primary adrenal carcinoma from adenomas. An adrenal to liver maximum standardized uptake value (SUV) ratio less than 1.45 was highly predictive of a benign lesion (sensitivity 100 %, specificity 88 %) [8].
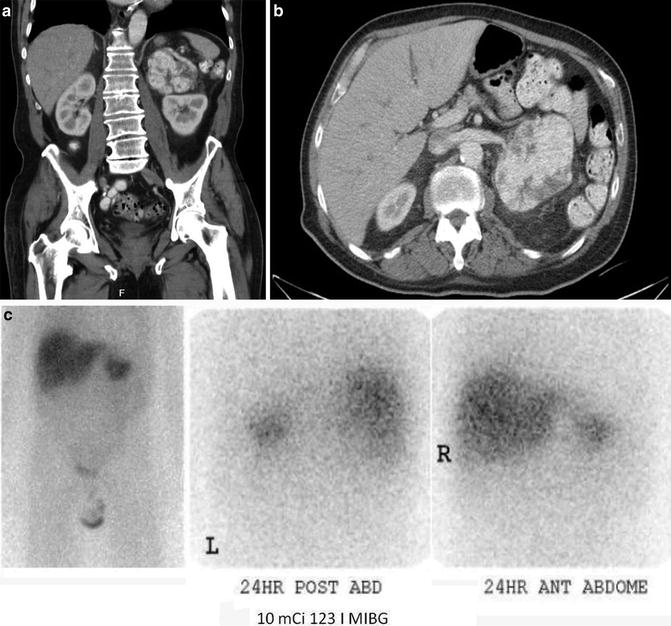
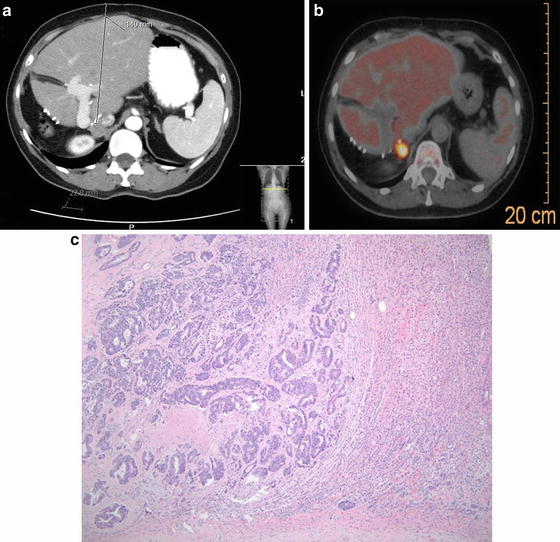
Fig. 14.4
A patient with a large left adrenal pheochromocytoma (a & b): CT scan reveals a large adrenal mass. (c): MIBG scan performed whole-body planar scintigraphic images of I-123 MIBG scan of a patient shows intense uptake in adrenal pheochromocytoma on the left side. The scan did not identify any other sites of disease with normal physiologic distribution of the radiotracer elsewhere in the body
Fig. 14.5
The patient is a 60-year-old with history of a colectomy and right hepatectomy in 2004. She presented with a solitary metastasis to the right adrenal. A biopsy proved to be a metastatic colon cancer. She underwent a successful resection in fall of 2013. (a): CT scan shows a mass in the right adrenal gland and a postoperative changes from a right hepatectomy. (b): PET scan reveals only uptake in the right adrenal gland. (c): Histology – image shows a focus of metastatic colorectal adenocarcinoma involving the adrenal gland. Benign adrenal parenchyma is seen on the right and a metastatic, moderately differentiated adenocarcinoma on the left. The tumor shows obvious glandular formation and areas of “dirty” necrosis, typical of colorectal adenocarcinoma (hematoxylin and eosin stain, original magnification, 100×)
Initial Work-Up and Evaluation of Hormonal Function
Majority of adrenal incidentalomas are benign, nonfunctioning adenoma; however, hormonal evaluation can reveal a significant number of patients of having unsuspected adrenal secreting lesions. Adrenalomas ≥1 cm and without prior history of malignancy should undergo hormonal evaluation. All patients with adrenal incidentaloma should be evaluated for possible pheochromocytoma, primary aldosteronism (hypertension, hyperkalemia), and Cushing’s syndrome (hypercotisolism) (Table 14.1). Approximately 5–7 % of adrenal incidentalomas are clinically silent pheochromocytoma [1–3, 6, 9, 10]. Primary aldosteronism should be considered in patients with hypertension and/or hypokalemia, although normokalemia can occur in up to 50 % of patients with hyperaldosteronism [11]. However, the diagnosis of primary aldosteronism can virtually be excluded in the absence of hypertension. Thus, laboratory testing should begin not only with the most sensitive tests but also with those that are the easiest to perform and the least expensive for the patient [3, 7].
Table 14.1
Evaluation of adrenal masses
Diagnosis | Images | Biochemical listing |
|---|---|---|
Primary aldosteronism (aldosteronoma) | CT/MRI | K+ <3.2; increased plasma aldosterone with decreased renin |
Pheochromocytoma | CT/MRI | 24-h urine metanephrines; plasma catecholamines |
MIBG scan | ||
Cushing’s syndrome | CT/MRI | 1 mg dexamethasone suppression test/ |
NP-59 scan | 24-h urine cortisol; plasma ACTH | |
Adrenal cortical carcinoma | CT/MRI | K+; 1 mg dex-suppression; 24-h urine free cortisol; 17-ketosteroids; and catecholamines |
Sex-steroid adenoma | CT/MRI | 17-ketosteroids; 24-h urine free cortisol |
Pheochromocytoma
Approximately 4.7 % of adrenalomas are silent pheochromocytoma, although some studies quote the incidence can be as high as 20 % [12, 13]. Almost 1/3 of all pheochromocytomas are discovered incidentally, and this prevalence increases over the span of time [14–16]. In an Italian retrospective multicenter study, 40 % of 234 pheochromocytomas were diagnosed between 1978 and 1997, while the remaining majority (59 %) were diagnosed in the last 5 years of the study [15]. Of note, prior to 1985 when utilization of ultrasound was limited, less than 10 % of pheochromocytomas were incidentally diagnosed [17]. Thus, the increase prevalence in pheochromocytoma may be a function of earlier detection. Of note, patients with asymptomatic pheochromocytomas tend to be older than those with symptomatic pheochromocytomas [3, 16, 17].
Some authors found that incidentally found pheochromocytomas tend to be large [3, 16]. By contrast, others found no size differences between incidentally found pheochromocytoma and symptomatic pheochromocytomas; however, they did find that patients with clinically silent pheochromocytoma had lower plasma catecholamine levels than those with symptoms [17].
Pheochromocytomas can be lethal even when they are clinically silent [3, 14]. Clinical presentation can vary, ranging from patients being entirely asymptomatic to those with intermittent headaches, palpitations, and sweating or with a hypertensive crisis. Patients may also be normotensive, which is not uncommon in cases where the pheochromocytoma was incidentally detected. In a multicenter study, Mantero et al. found that about 50 % of patients with incidentally detected pheochromocytoma were normotensive, while the others had mild to moderate hypertension. None of the patients had paroxysmal symptoms of adrenergic excess [9]. Because a significant percentage of patients with pheochromocytoma can be asymptomatic and normotensive, any patient with an incidental adrenaloma should undergo biochemical testing for pheochromocytoma [3]. Outcome for patients with malignant pheochromocytoma is poor; the mean 5-year survival rate is about 40 % [18].
Biochemical Diagnosis of Pheochromocytoma
The diagnosis of pheochromocytoma has evolved over the last six decades. It became apparent in the very early century that the clinical signs and symptoms of pheochromocytoma were due primarily to the excess secretion of catecholamines. In the middle of the last century, a 24-h urinary catecholamines excretion became the test of choice to account for the episodic nature of catecholamines secretion [19–21]. In recent years, metabolites of catecholamine such as normetanephrine and metanephrine, which are respective o-methylated metabolites of norepinephrine and epinephrine, and vanillylmandelic acid (VMA), the final breakdown product of the catecholamines was included as part of the biochemical diagnostic tests [19].
There is no consensus on what the preferred test for pheochromocytoma should be [3, 4, 12, 22]. Recently, Lenders et al. assessed 858 patients with adrenalomas who were at risk for pheochromocytoma [23]. Patients were identified as having a pheochromocytoma based on signs and symptoms suggestive of a pheochromocytoma, or on their genetic predisposition to developing pheochromocytoma. Plasma and urinary catecholamines, urinary fractionated metanephrine and VMA, as well as plasma values of free metanephrine were measured. Findings from this study demonstrated that plasma free metanephrine was the optimal screening test for pheochromocytoma, yielding a 99 % sensitivity and 89 % specificity. The false-negative rate was very low with only 3/214 patients with pheochromocytoma having negative laboratory values [23]. However, plasma metanephrine testing may not be readily available in some centers; therefore, a 24-h urinary metanephrines remains an acceptable initial screening test. The degree of increase in catecholamines and metanephrines can be useful for diagnostic and therapeutic purposes since a mild increase may not necessarily be due to a pheochromocytoma but be due to other causes such as diet and pharmacologic causes [24].
Hereditary Pheochromocytoma and Paraganglioma
Pheochromocytoma and paraganglioma (pheochromocytomas that are outside of the adrenal glands) are neuroendocrine tumors that are derived from sympathetic and parasympathetic paraganglioma. The most common extra-adrenal site of pheochromocytoma is the organ of Zuckerkandl, which comprises of small masses of chromaffin cells along the aorta, with the highest concentration located at the origin of the inferior mesenteric artery and the bifurcation of the aorta.
Most leaders in the field would agree that the rule of 10 applies to pheochromocytoma: 10 % are hereditary (Fig. 14.6), 10 % are bilateral, 10 % are extra-adrenal (Fig. 14.7), 10 % are malignant (Fig. 14.2), and 10 % occur in children. Pheochromocytomas of adrenal and extra-adrenal sympathetic origin usually secrete catecholamines, whereas lesions of parasympathetic origin (head & neck) usually do not [14]. Roughly one third of pheochromocytomas have germline mutation, and approximately ten tumor susceptibility genes have been identified [25]. These include SDHA/B/C/D (succinate dehydrogenase complex subunits A, B, C, & D), SDHAF2 (succinate dehydrogenase complex assembly factor-2), VHL (von Hippel–Lindau) (Fig. 14.6), RET (REarranged during Transfection), NF1 (neurofibromatosis type 1), and recently reported TMEM127 (transmembrane protein 127) and MAX (Myc-associated factor X) [25, 26]. Somatic mutations in RET, MAX, VHL, and NF1 have been reported in 17 % of sporadic lesions [26–29]. Succinate dehydrogenase (SDH) is an important part of the mitochondrial electron transport chain, and when it is mutated, the ability of cells to phosphorylate is undermined [30–33].
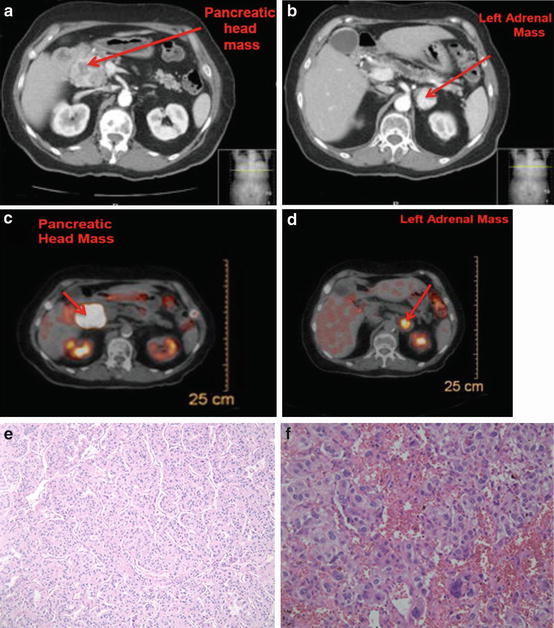
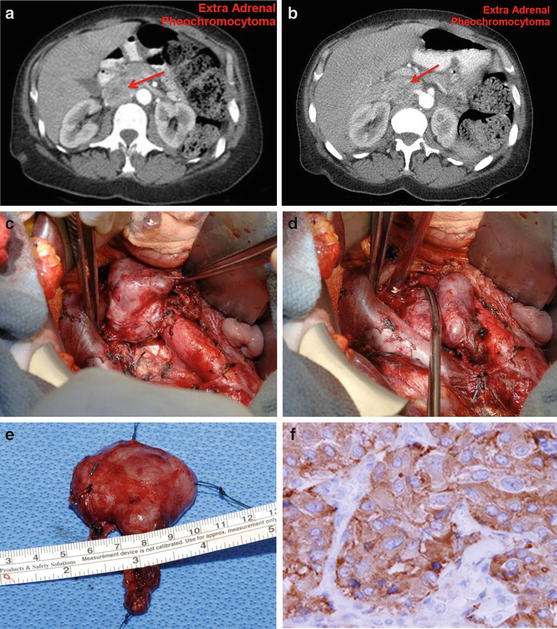
Fig. 14.6
A patient with von Hippel–Lindau disease with a pancreatic head mass and a left adrenal mass. She underwent a left adrenalectomy (pheochromocytoma) and a Whipple procedure (neuroendocrine tumor of the pancreas). (a): CT shows a mass in the head of the pancreas (neuroendocrine tumor) (b): CT shows a left adrenal mass (pheochromocytoma) (c): PET scan shows positive uptake in the head of the pancreas (d): PET scan shows positive uptake in the left adrenal gland as well as in the head of the pancreas. (e): Histology – this photomicrograph depicts a pancreatic endocrine neoplasm. This tumor manifests a typical trabecular pattern of uniform cells with a granular eosinophilic cytoplasm and round nuclei with finely granular, “salt-and-pepper” chromatin pattern (hematoxylin and eosin stain, original magnification 200×) (f): This photomicrograph of a pheochromocytoma illustrates the typical “Zellballen” architectural pattern in which balls of tumor cells are supported by a rich vascular framework. The cytoplasm has a finely granular appearance, and nuclei demonstrate a stippled chromatin pattern. Though mitotic activity is typically sparse in these tumors, note the enlarged and hyperchromatic nuclei (hematoxylin and eosin stain, original magnification 200×)
Fig. 14.7
Extra-adrenal pheochromocytoma (organ of Zuckerkandl) (a & b): CT – Arrow points to a retroperitoneal mass, which is inferior to the superior mesenteric artery and between the inferior vena cava, aorta, and left renal vein. (c & d): Intraoperative photos of extra-adrenal pheochromocytomas located between the inferior vena cava and aorta and below the left renal vein. (e): Gross picture (f): Histology – the tumor shows nests and trabecular composed of large polygonal cells with finely granular eosinophilic to somewhat basophilic cytoplasm. There is mild nuclear pleomorphism, and the nucleoli are small and eccentric. The tumor cells reveal positive cytoplasm stain for chromogranin and synaptophysin. Common sites for extra-adrenal pheochromocytomas: between the inferior vena cava and aorta below the left renal vein, and in the organ of Zuckerkandl. Small tumors under the left renal vein may be overlooked unless the area is carefully inspected
Preoperative and Intraoperative Management of Pheochromocytoma
The treatment of choice for pheochromocytoma is surgical excision. In the past, poor outcomes following resection of pheochromocytomas were due to uncontrolled hypertensive crisis related to excess catecholamine secretion. The first report of surgical excision of a pheochromocytoma in North America was performed by Dr. C. Mayo in 1927 [34]. In the 1950s, the group at the Mayo Clinic reported a decreased operative mortality with successful resection of 61 pheochromocytoma over 11 years by using alpha-blockade with phenoxybenzamine to treat hypertension and epinephrine to treat intraoperative hypotension [35].
However, before surgical intervention, perioperative management requires adequate hydration with isotonic solution and administration of selective alpha 1-adrenergic blocking agents (i.e., doxazosin, prazosin, or terazosin) followed by beta-adrenergic blockade (i.e., propranolol, atenolol) (Table 14.2). It is very important that beta-blockade should never be initiated first because blockade of the vasodilatory peripheral beta-adrenergic receptors with unopposed alpha-adrenergic receptors stimulation can lead to a further elevation of blood pressure.
Table 14.2
Drugs used to treat pheochromocytoma
Drug | Initial dose | Maximum dose | Suggestion |
|---|---|---|---|
Phenoxybenzamine | 10 mg orally twice daily | 2 mg/kg/day | Nonselective alpha-blockade may titrate to 3 times per day until postural hypotension is achieved |
Prazosin | 1 mg orally twice daily | 15 mg/day | Selective alpha-blockade, same as above |
Labetalol | 100 mg orally twice daily | 1,200 mg/day | After alpha-blockade, may titrate the dose with target heart rate of 60–70 bpm |
Nifedipine | 30–90 mg orally once daily | 120 mg/day | Calcium channel blocker may increase after 1 week, used for paroxysmal hypertension |
Amlodipine | 5 mg | 10 mg/day | Same as nifedipine |
Metyrosine | 250–750 mg orally four times daily | 4,000 mg/day | Used for refractory hypertension |
Calcium channel blockers (i.e., nifedipine) are alternative vasodilator to control blood pressure. These drugs are necessary to normalize heart rate and blood pressure. As mentioned before, it is necessary to replete intravascular volume preoperatively and take precautionary measures to prevent cardiovascular collapse from surgery-induced catecholamine storm [22, 36, 37]. Even with proper and effective preoperative alpha- and beta-adrenergic blockade, hypertensive crisis may occur intraoperatively due to catecholamine surge secondary to manipulation of the adrenal lesion. It is very important for the patient to have an arterial line and a central venous line placed. Additionally, the anesthesiologist must have ready-at-hand premixed intravenous titratable drugs to control blood pressure in the event the blood pressure fluctuates during surgery. Dihydropyridine calcium channel blocker nicardipine (0.5–1.0 mg/ml) and the direct-acting vasodilator sodium nitroprusside (0.5–3 μg/kg/min) are the agents of choice for uncontrolled hypertension. On the other hand, ligation of the adrenal vein can cause an abrupt decrease of catecholamine release, which can lead to unexpected hypotension. Should this occur, epinephrine or norepinephrine should be administered accordingly, along with crystalloid infusion as needed [15, 22, 38].
Operative Management and Technique
Adrenal lesions can be removed by an open technique or by a minimally invasive technique, either via a transabdominal or retroperitoneal approach. Adrenalectomy is effective at treating a number of different diseases and conditions (Table 14.3) including but not limited to pheochromocytoma, aldosteronoma, refractory Cushing’s disease, adrenal cortical carcinoma, and selected cases of metastatic disease confined to the adrenal gland. Until 1992 when the first laparoscopic adrenalectomy was reported in the literature by Ganger et al., the only operative approach for removal of the adrenal gland was an open adrenalectomy [37]. Laparoscopic and more recently robotic adrenalectomy techniques have been considered by many to be the “gold standard” and the procedure of choice for resecting adrenal lesions. In experienced hands, these techniques are safe and can lead to less postoperative pain, shorter hospital stay, better cosmesis, and less blood loss [37–42]. Open surgery is generally reserved for large lesions, especially those with malignant features and locally advanced. The retroperitoneal approach might be an attractive option for patients who have multiple prior abdominal surgeries. On the other hand, transabdominal approach has the advantage of addressing other intra-abdominal pathology as well as contralateral adrenal lesion. Therefore, most leaders in the field chose the surgical procedure based upon whether the adrenal mass is hereditary or sporadic and whether the lesion is unilateral or bilateral. More importantly, the surgeon should choose the technique for which he or she has the most experience and comfort.
Table 14.3
Indications for adrenalectomy
Hormonally active |
Aldosteronoma |
Glucocorticoid adenoma |
Pheochromocytoma |
Bilateral macronodular adrenal hyperplasia |
Selected cases of bilateral adrenal hyperplasia due to Cushing’s disease or ectopic ACTH syndrome |
Adrenocortical carcinoma |
Hormonally inactive |
Adrenal carcinoma |
Metastatic disease with controlled primary site/otherwise biopsy and systemic treatment |
Tumors >5 cm |
Tumors that demonstrate growth on serial imaging |
If a patient presents with bilateral hereditary disease, the surgeon may consider performing a unilateral cortex-sparing adrenalectomy on one side and a contralateral total adrenalectomy for pheochromocytoma. For patients with a metachronous contralateral pheochromocytoma after prior unilateral adrenalectomy, a cortex-sparing procedure should be attempted [38, 43].
Adrenal Carcinoma (ACC)
Adrenal cortical carcinoma (ACC) is a very rare cancer that accounts for 1 % of all adrenal lesions. It affects two person per million, worldwide [44]. It is the second most lethal endocrine tumor after anaplastic thyroid malignancy [45]. There is a bimodal age distribution with a peak occurrence in the first decade of childhood and another in the fourth and fifth decades of life. The female to male ratio is approximately 1.5–1.0 [46]. The majority of these cases occur in a sporadic fashion, although ACC can occur in association with several hereditary syndromes including Li–Fraumeni syndrome, Beckwith–Wiedemann syndrome, and multiple endocrine neoplasia syndrome type 1 (MEN-1) [46, 47]. The prognosis of patients with this cancer remains very poor, despite its earlier detection by modern imaging modalities.
Related posts:
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


