The incidental finding of one or more adrenal masses, discovered on a radiological study performed for reasons other than suspected adrenal disease, has become a common occurrence in modern medical practice. Because the differential diagnosis for these masses, termed adrenal incidentalomas, incudes multiple distinct clinical entities (of differing prognostic significance), surgical oncologists must be familiar with the appropriate diagnostic workup and management of these lesions. This requires not only an understanding of adrenal anatomy, pathophysiology, biochemistry, genetics, and differential diagnosis, but also a working knowledge of the upstream hypothalamic-pituitary axis, which regulates adrenal function.
The prevalence of adrenal incidentalomas depends on the resolution of the imaging modality used, with the most sensitive computed tomography (CT) scanners identifying unexpected adrenal masses in up to 4% of abdominal scans.1,2 Prevalence is also proportional to age, varying from approximately 1% among individuals less than 30 years old to 10% in those over 70.3–5 Bilateral lesions are present in 10% to 15% of cases and autopsy series indicate equal prevalence among men and women. Adrenal incidentalomas are also more common among patients diagnosed with hypertension, diabetes, and/or obesity.1,3,5,6
The workup of a newly diagnosed adrenal incidentaloma begins with a thorough history and physical exam, with emphasis placed on two questions: (1) Is excess adrenal hormone production present? and (2) Is primary malignancy or metastatic disease present?
Abdominal and/or flank pain resulting from the development of an adrenal mass is unusual and, when present, often connotes an adrenal malignancy and poor prognosis. These symptoms may also be limited to large adrenal masses, usually >6 cm in diameter, or to masses for which rapid enlargement has occurred (usually the result of intralesional hemorrhage). The majority of adrenal incidentalomas do not produce abdominal pain, regardless of their size.
With the exception of metastatic disease involving the adrenal gland(s), all adrenal incidentalomas represent abnormal growth of adrenal tissue. As such, all the patients must be evaluated for associated overproduction of adrenal hormones, including catecholamines (pheochromocytoma), cortisol (Cushing’s syndrome and subclinical Cushing’s syndrome), aldosterone (Conn’s syndrome), and sex steroids (virilization or feminization—usually associated with adrenocortical carcinoma). This assessment begins with careful evaluation for the presence of associated symptoms (Table 41-1) and is followed by biochemical testing.
Signs and Symptoms of Excessive Hormone Production in Patients Presenting with an Adrenal Incidentaloma
| Cushing’s Syndrome | Pheochromocytoma | Aldosteronoma |
|---|---|---|
| Hyperglycemia/diabetes | Headache | Hypertension |
| Osteoporosis | Palpitations/tachycardia | Headache |
| Hypertension | Hypertension | Hypokalemia |
| Weight gain | Diaphoresis | Hypocalcemia |
| Atherosclerosis | Anxiety/panic attacks | Myalgia and muscle spasm |
| Tremor | ||
| Weakness |
In addition to hormonal testing, all patients diagnosed with an adrenal incidentaloma should undergo CT scanning. The primary purpose of this testing is to estimate the probability that a given incidentaloma represents a malignancy and, if malignancy is present, to assess neighboring tissues for possible invasion. In addition, the contralateral adrenal gland can be evaluated for concurrent abnormalities. Secondarily, CT scans can help elucidate whether or not the patient has a pheochromocytoma with its distinctive intravenous washout characteristics.
As the size of an adrenal lesion increases, the associated risk of malignancy also increases; those tumors less than 4 cm are almost never malignant, whereas tumors initially identified as greater than 4 cm have a proportionally increased risk of being malignant. It is, therefore, recommended that surgical resection be performed for all adrenal tumors 4 cm or greater regardless of CT scan characteristics.7–12 Because adrenal incidentaloma size is proportional to associated malignancy risk, multiple investigators have surmised that the growth of nonfunctional incidentalomas, identified on serial imaging, might also predict malignancy. This correlation has not, in itself, proven to be 100% sensitive or specific, and incidentaloma growth should not be considered in isolation when evaluating for surgical resection.13
Adrenal protocol CT scanning is the preferred imaging modality for assessment of adrenal incidentalomas. This technique includes multiple imaging phases, including noncontrast (unenhanced phase) images, images obtained 1 minute after IV contrast administration (enhanced phase) and images acquired 10 to 15 minutes after IV contrast is given (washout phase). This protocol also uses fine-cut resolution (2.5 to 3 mm reconstructions) to accurately define incidentaloma size. The cells of benign adrenal lesions are, in most cases, characterized by a high cytoplasmic fat content, which manifests as relatively low attenuation on unenhanced CT imaging. Regardless of diameter, an enhancement value of less than 10 Hounsfield units (HU) on noncontrast CT scanning is 100% specific for benign disease and, if nonfunctional and less than 4 cm, surgical resection should be deferred.14 Similarly, identification of intralesional macroscopic fat on CT scanning is pathognomonic for myelolipoma, an inherently benign adrenal tumor (Fig. 41-1). Lesions in which unenhanced imaging yields HU greater than 10 but a washout of 50% or greater (comparing the enhanced phase to the washout phase) are almost always benign and, if nonfunctional, do not require surgical resection (Fig. 41-2).15,16 Nonfunctional adrenal incidentalomas that do not meet these imaging criteria should be referred for surgical resection (Fig. 41-3).
FIGURE 41-1
Unenhanced CT imaging of a right-sided adrenal myelolipoma. A 55-year-old woman presented with a 5.2-cm right adrenal mass (red arrow) discovered incidentally on MRI evaluation for spinal stenosis. A biochemical evaluation for excess adrenal hormone production was negative. The mass demonstrated multiple foci of macroscopic fat, an example of which is shown (white arrow, HU -83). This finding is pathognomonic for myelolipoma, a benign lesion.
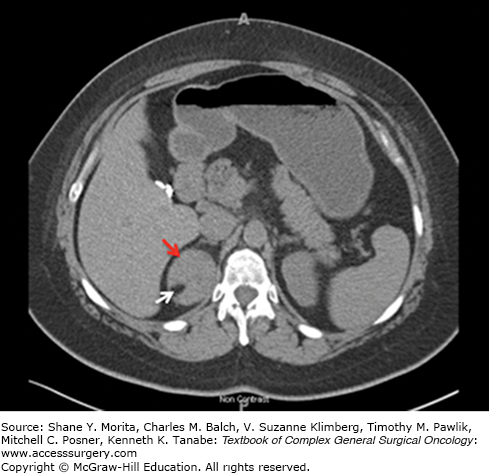
FIGURE 41-2
CT imaging of a nonfunctional right adrenal adenoma. A 35 year-old woman presented with a 3.4 cm right adrenal mass (red arrows) discovered incidentally on CT scanning during a trauma work-up following a motor vehicle accident. Biochemical evaluation for excess adrenal hormone production was negative. The mass demonstrated an unenhanced attenuation of 18 HU (left panel), an enhanced attenuation of 91 HU (middle panel), and a washout attenuation of 30 HU (right panel). The percentage drop in attenuation observed when comparing enhanced to washout images is 67%, consistent with a benign lesion.
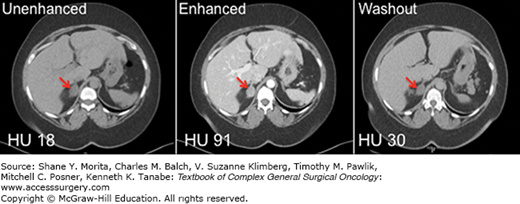
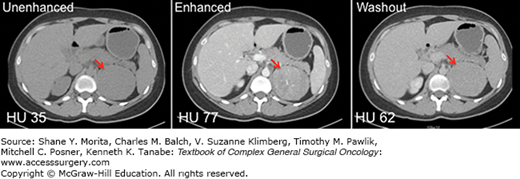
FIGURE 41-3
CT imaging of a left-sided adrenocortical carcinoma. A 27-year-old woman presented with an 8-cm left adrenal mass (red arrows) discovered incidentally on CT evaluation for a history of polycystic ovarian syndrome. A biochemical evaluation for excess adrenal hormone production was negative. The mass demonstrated an unenhanced CT attenuation of 35 HU (left panel), an enhanced attenuation of 77 HU (middle panel), and a washout attenuation of 62 HU (right panel). The percentage drop in attenuation observed when comparing enhanced and washout images is 19%, consistent with malignancy.
Chemical shift magnetic resonance imaging (CS-MRI) can also be used to assess adrenal incidentaloma malignancy risk, particularly when CT scanning is unavailable or contraindicated, and reported sensitivities and specificities for this technique range between 81% to 100% and 94% to 100%, respectively.17,18 Positron emission tomography (PET) scanning is generally not recommended in the assessment of incidentaloma malignancy risk, as most PET scanners provide inadequate spatial resolution (small lesions may be missed) and are associated with unacceptably high false-positive rates (up to 13%).17,19,20
Fine-needle aspiration (FNA) biopsy plays a very limited role in the diagnostic workup of adrenal incidentalomas. This technique is only useful in identifying metastatic disease involving the adrenal glands, as primary adrenal malignancies are generally indistinguishable from benign adrenal tissue on cytologic analysis. Aspiration biopsy may also result in seeding of adrenocortical carcinoma cells to surrounding tissues and thus should not be performed if primary adrenal malignancy is suspected.21,22 Most important, FNA biopsy of a pheochromocytoma may result in life-threatening hypertensive crisis or significant gland hemorrhage.23 For all of the above reasons, adrenal FNA should only be considered when metastatic disease to the adrenal gland(s) is suspected and no other lesions amenable to biopsy are present. Pheochromocytoma must also be ruled out prior to biopsy and the probability of a primary adrenocortical carcinoma must be low.
Pheochromocytomas are derived from chromaffin cells of the adrenal medulla and account for approximately 3% of adrenal incidentalomas.24 The classic triad of symptoms associated with pheochromocytoma includes headache, diaphoresis, and palpitations, although the majority of pheochromocytoma patients will not experience all three symptoms (Table 41-1). Hypertension, often refractory to medical therapy, is the most common sign produced by a pheochromocytoma. Nonetheless, hypertension will not be present in up to 50% of patients with an incidentally discovered pheochromocytoma.25 Careful assessment of family history is critically important when pheochromocytoma is suspected, as pheochromocytoma is a known feature of several predisposing familial syndromes, including multiple endocrine neoplasia type 2A, type 2B, Von Hippel–Lindau syndrome and neurofibromatosis type 1.
Because of the significant potential for severe morbidity associated with an undiagnosed pheochromocytoma, including hypertensive crisis, cardiac failure, and stroke, all patients diagnosed with an adrenal incidentaloma should undergo biochemical testing for pheochromocytoma. The ideal biochemical test remains controversial. Some experts prefer assessment of fractionated catecholamine, metanephrine, normetanephrine, and dopamine levels in a 24-hour urine collection. This affords a diagnostic sensitivity and specificity of 98%, but is inconvenient for the patient and potentially inaccurate unless strict collection compliance is maintained.26,27 In addition, interfering medications, which include psychoactive agents (other than selective serotonin reuptake inhibitors), levodopa, alpha-blockers, and ethanol, must be discontinued a minimum of two weeks prior to performing this assay.28 A simpler test is assessment of plasma fractionated metanephrines, which is associated with a 96% to 100% diagnostic sensitivity but only 89% specificity. Thus, the primary utility of this test is identification of patients who do not have pheochromocytoma, as a negative test virtually rules out all but very rare dopamine-only secreting tumors.29 In contrast, a positive test should prompt confirmation by 24-hour urine assessment.
Pheochromocytomas almost always demonstrate a HU attenuation greater than 10 on unenhanced CT imaging and, when comparing enhanced to washout CT scan phases, an enhancement decrement of less than 50% (which may lead to misinterpreted as an adrenocortical carcinoma) (Fig. 41-4). The classic MRI finding is “light bulb” bright intensity on T2-weighted imaging, relative to cerebrospinal fluid signal intensity, while T1 imaging tends to demonstrate isointensity relative to the liver.17 Two-thirds of pheochromocytomas will be solid, with the remaining proportion being cystic or complex. Calcifications will be present in up to 21% of cases and hemorrhage within a pheochromocytoma may produce heterogeneous internal consistency on CT imaging.30,31 Bilateral disease will be present in approximately 10% of cases and 10% of pheochromocytomas will be malignant.
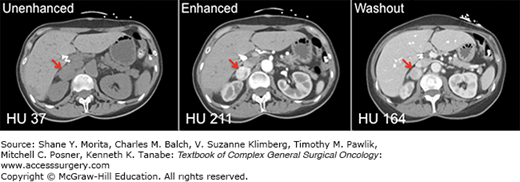
FIGURE 41-4
CT imaging of a right-sided pheochromocytoma. A 57-year-old woman presented with complaints of severe headaches, tachycardia, and refractory hypertension. A 24-hour urine assay demonstrated marked elevation in catecholamine, metanephrine, and normetanephrine levels. A 3.1-cm right adrenal mass was demonstrated on CT imaging (red arrows), with associated unenhanced attenuation of 37 HU (left panel), enhanced attenuation of 211 HU (middle panel), and washout attenuation of 164 HU (right panel). The percentage drop in attenuation observed when comparing enhanced to washout images is 22%.
Malignant pheochromocytomas are associated with mutation in the succinate dehydrogenase B gene and patients carrying this mutation are more likely to develop malignant pheochromocytomas.32 Malignancy cannot be distinguished from benign disease on histologic analysis; only the presence of local invasion and/or distant metastases is indicative of cancer, and malignancy may not become apparent for up to 20 years following initial resection.33,34 For this reason, all patients having undergone adrenalectomy for apparently benign pheochromocytomas should be evaluated by annual plasma fractionated metanephrine testing, as recurrent or metastatic disease will develop in up to 16% of cases.35
All patients diagnosed with a pheochromocytoma should be referred for unilateral adrenalectomy or, when bilateral disease is present, cortical sparing adrenalectomy. Alpha adrenergic blockade, most commonly achieved using the long acting, nonspecific alpha-blocker phenoxybenzamine, should be initiated in all cases 2 or 3 weeks prior to surgery in order to prevent potentially life-threatening intraoperative hemodynamic instability. Initial dosing of phenoxybenzamine should be 10 mg, given twice daily, and this dose should be titrated up until normotension is achieved or significant side effects develop, such as nasal congestion, tachycardia, abdominal pain, nausea, and/or orthostatic hypotension. Intravascular volume expansion should be concurrently initiated to compensate for the relative volume depletion associated with excess catecholamine production, either with intravenous normal saline or by increased oral intake of salt and water. Beta blockade should not be initiated for at least 2 days after alpha-blocker therapy has begun so as to avoid the unopposed alpha stimulation, and potential hypertensive crisis, associated with beta-blocker monotherapy in these patients. Postoperative antihypertensive medication dosing should be titrated to blood pressure and, if essential hypertension is absent, should be discontinued. Alpha blockade should be continued for symptomatic control in patients having undergone debulking only or incomplete resection and should be restarted in cases of recurrence.11
Related posts:




Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


