Mild hypercortisolism
Overt hypercortisolism
Diagnostic tests
Sensitivity/specificity
DSTa
75–100%/67–72%
100%/91%
UFCb
36%
97%/91%
MSCc
82%/60%
92–100%/93–100%
Frequent mutations d
Genetic mutations
CTNNB1 (16%)
ARMC5 (55% of BMAH)e
PRKACA (35–69%)
GNAS (5–17%)
PRKAR1A (66% of PPNAD)f
It is now understood that while some gene mutations may primarily lead to tumor growth, others may prominently lead to excessive steroidogenesis [19]. As epidemiological data has shown that the majority of cases of mild hypercortisolism do not progress to overt disease, it has been hypothesized that some mutations might preferentially lead to subclinical disease, while others lead to greater cortisol overproduction and the typical phenotype of overt Cushing’s syndrome [15, 17].
In the case of adrenocortical tumors, most seem to arise in a sporadic manner, with a minority of cases manifesting in the setting of neoplasia-prone hereditary syndromes (e.g., multiple endocrine neoplasia 1, McCune-Albright syndrome, and Carney complex) [20]. Mutations at various levels of the cAMP/protein kinase A (PKA) signaling pathway have recently been singled out as major contributors to adrenal Cushing’s syndrome. The most common somatic activating mutations in these adenomas are those found in the catalytic subunit of PKA (PRKACA gene) and the G-protein alpha subunit (GNAS gene). These mutations have been reported in up to 35–69% and 5–17% of all sporadic adrenocortical adenomas causing overt Cushing’s syndrome, respectively [21–23].
Aside from mutations affecting the cAMP/PKA pathway, alterations in Wnt β-catenin signaling have also been reported frequently in cortisol-secreting adenomas. Sixteen percent of studied adenomas in a tissue repository were found to harbor mutations in the CTNBB1 gene, which is responsible for encoding β-catenin [24].
Tumors that contain the PRKACA and GNAS mutations tend to be smaller and linked to overt disease, while neoplasms exhibiting mutations in the CTNBB1/Wnt β-catenin have more ineffective steroidogenesis while favoring tumoral growth. This phenomenon was confirmed in an exome sequencing study of 25 tumor-normal pairs, as only adenomas with PRKACA/GNAS mutations were linked to overt disease and earlier diagnosis (13/16 with PRKACA or GNAS mutation with overt hypercortisolism versus 16/39 without, p = 0.008). Of note, PRKACA/GNAS and CTNBB1 mutations appeared to be mutually exclusive, supporting the hypothesis of a distinct genetic precedent between the different phenotypes of mild vs. overt adrenal hypercortisolis m [25].
Diagnosis
According to the 2008 Endocrine Society guidelines for the diagnosis of Cushing’s syndrome, a thorough medical history and physical exam should be performed on all subjects suspected of having endogenous hypercortisolism. Special attention should be paid to any possible exogenous steroid exposure, as glucocorticoid intake remains the most common cause of pathologic hypercortisolism. Additionally, reviewing the patient’s medications is crucial, as drugs that either induce or inhibit CYP3A4 metabolism (e.g., certain antiepileptics or itraconazole, respectively) or increase cortisol-binding globulin (such as estrogen-containing contraceptives) may lead to false-negative or false-positive results [1].
Three tests may be used to make the diagnosis of hypercortisolism in general: the low-dose dexamethasone suppression test, a 24-hour urinary free cortisol, and a midnight salivary cortisol. These tests aim to identify the evidence of failure to suppress cortisol production in spite of ACTH suppression, the presence of excess production of cortisol, and the loss normal diurnal variation in cortisol secretion, respectively (see Table 10.1).
The low-dose dexamethasone suppression test (LDDST) is the standard exam used for ruling out hypercortisolism from any cause. When using a LDDST, suppression of the plasma cortisol level to <1.8 μg/dL has the best negative predictive value and overall best sensitivity to rule out hypercortisolism (100%) [26].
Several dexamethasone suppression tests are available for confirming the presence of hypercortisolism (with either a low-dose 1 mg overnight test or the 2-day 2 mg test). The 2-day, 2 mg LDDST has been postulated as a superior screening tool for Cushing’s syndrome. The test is performed by taking 0.5 mg of oral dexamethasone every 6 h (usually starting at 8 a.m.) and measuring serum cortisol and dexamethasone at 8 a.m. on day 3. A retrospective study of 245 patients with pituitary Cushing’s syndrome found the test to correctly identify hypercortisolism in 98% of cases [27]. Although this test retains excellent sensitivity while exhibiting better specificity when compared to the standard LDDST, it requires greater effort from the patient, as the frequency of dosing may be too cumbersome for some. Being a synthetic corticosteroid, dexamethasone does not interact with the antibody immunoassays currently in use and is therefore the glucocorticoid of choice when trying to suppress endogenous cortisol production.
Infrequently, dexamethasone may be poorly absorbed or excessively metabolized, such as in the case of concomitant use of CYP3A4 altering drugs (inducers such as rifampicin and anticonvulsants, inhibitors such as protease inhibitors and azole antifungals) [28]. The simultaneous measurement of the dexamethasone level during LDDST is critical in order to verify adequate serum concentrations. An ideal serum dexamethasone level should be ~200–650 ng/dL [29]. Hormonal contraceptive use in women also requires special consideration, as estrogen has been found to increase cortisol-binding globulin (CBG) levels and therefore potentially raise total cortisol levels and render the dexamethasone suppression test uninterpretable [30]. It is highly recommended to postpone the biochemical evaluation of adrenal hyperfunction until after cessation of hormonal contraceptives, given the risk of false-positive results.
The excellent negative predictive value of the LDDST comes an increased risk of false-positive results; therefore, a positive test should be followed by additional confirmatory testing, including the collection of 24 h urine free cortisol and a midnight salivary cortisol assay. Given the variable nature of Cushing’s syndrome, two measurements of each of these tests are recommended when confirming the diagnosis.
The use of 24-hour urine free cortisol (UFC) measurement is a unique way of quantifying global glucocorticoid exposure in the tissues, primarily reflecting serum cortisol not bound to CBG or other proteins. This is ideal under conditions where CBG abnormalities may be suspected, such as during pregnancy where higher CBG levels are to be expected or during conditions with protein wasting such as the nephrotic syndrome, where lower CBG levels might lead to false-negative results. Structure-based analysis such as tandem mass spectrometry is preferred over immunoassays, as this eliminates the risk of interference from cortisol precursors and metabolites [31].
Care must be taken when interpreting mildly elevated 24 h UFC values (<2 times the upper limit of normalcy), as false-positive results may be seen in conditions such as pseudo-Cushing’s or physiologic hypercortisolism [32]. Other disorders such as renal insufficiency, polydipsia (water intake >5 L daily), or polyuria (urine output >3 L over 24 h) may also decrease the accuracy of the test; when present, other screening methods should be favored. A study by Mericq et al. on normal subjects showed that high fluid consumption caused the mean UFC values to increase (126 ± 33 (SD) vs. 77 ± 18 μg/day, p < 0.005). Additionally, more samples exceeded the upper limit of normalcy of 95 μg/day (23/30 vs. 6/30, p < 0.005) [33].
Midnight salivary cortisol testing is a convenient way of looking for the loss of diurnal variation in cortisol secretion, which can be easily performed in an outpatient setting. As with the other available tests for evaluating hypercortisolism, late-night salivary cortisol measurements may be influenced by factors such as age, sex, weight, and sleep habits of the individual as well as the presence of cyclical cortisol secretion in certain adenomas [34, 35].
Once endogenous hypercortisolism has been diagnosed biochemically, a morning ACTH should be measured. A suppressed level (<10 pg/mL) establishes independence from pituitary control and the absence of an ectopic ACTH source as the cause of hypercortisolism, confirming the diagnosis of an adrenal cause. However, in some patients with mild adrenal hypercortisolism, the degree of excess cortisol production may not be sufficient to suppress the hypothalamic-pituitary-adrenal (HPA) axis, and a low-normal morning ACTH level may be seen. In these cases, an ACTH can be checked after LDDST. If the ACTH level is not suppressed in the setting of a therapeutic dexamethasone level, other causes such as pituitary or ectopic ACTH overproduction should be considered.
Other labs may also be supportive of the diagnosis, including a low dehydroepiandrosterone (DHEA) sulfated (DHEA-S) level. A low DHEA-S (<40 μg/dL) has been found to have a sensitivity of 68%, specificity of 75%, PPV of 43%, and NPV of 90%. This decrease may be less clinically impactful in the elderly population, as serum DHEA and DHEA-S are known to decrease with aging [36].
Overt Hypercortisolism
Patients with overt Cushing’s syndrome typically exhibit clinical features that best discriminate from other conditions. In this setting of high pretest probability of disease, tests that favor a high degree of specificity over sensitivity are preferred as “rule-in” tools to diagnose hypercortisolism. The 24 h urine free cortisol (when obtained appropriately and multiple times) arguably has the best accuracy with more severe forms of cortisol excess, particularly when the value is higher than three times the upper limit of normal [1]. A meta-analysis looking at 646 individuals with confirmed disease found that a patient with an abnormal 24 hour UFC result had a high likelihood of having endogenous hypercortisolism (LR 10.6, 95% CI 5.5–20.5) [37].
There is also data demonstrating that midnight salivary cortisol measurements are very sensitive and specific for establishing the diagnosis of overt Cushing’s syndrome. In a study assessing the predictivity of the test in 13 patients with overt disease and 14 patients with mild hypercortisolism, late-night salivary measurements at a threshold of 12 nmol/L displayed a 100% sensitivity and specificity for the diagnosis of overt Cushing’s syndrome [38]. Of note, the cohort came from a mixed setting (both outpatient- and hospital-based measurements).
Given its ease of use and widespread availability, a LDDST still remains an essential exam to rule out pathological cortisol secretion. As mentioned previously, the test retains an excellent degree of sensitivity in the setting of overt cortisol excess. While ideal as a screening tool, LDDST carries some pitfalls. There have been retrospective reports of cortisol suppression to <2 μg/dL with established Cushing’s syndrome; a single center cohort examined by Findling et al. showed a minority (6 out of 80 patients) to exhibit this response to a LDDST [39].
Mild Hypercortisolism
As mentioned previously , controversies as to the biochemical definition and the diagnostic parameters needed to identify the presence of subclinical hypercortisolism remain. The LDDST has been shown to be the most sensitive test for the detection of mild adrenal hypercortisolism. The morning serum cortisol value after 1 mg dexamethasone the night before that is considered diagnostic of mild hypercortisolism has been debated; historically, literature suggested that >5 μg/dL (>138 nmol/L) after LDDST was diagnostic. However, more recently authors have proposed that the traditional threshold after LDDST, a morning cortisol of >1.8 μg/dL (>50 nmol/L), be employed to establish the diagnosis of mild hypercortisolism. There are others who suggest using a morning cortisol of >3 μg/dL (>80 nmol/L), a value to help balance between sensitivity and specificity. The discrepancy arises because of the difference in clinical index of suspicion between Cushing’s syndrome and mild hypercortisolism. In patients being screened for symptomatic Cushing’s syndrome, there is a high pretest probability of disease, so the likelihood of a false-positive result with a morning cortisol of >1.8 μg/dL is low. However, patients with mild hypercortisolism do not have the typical stigmata so the clinical index of suspicion is lower, and therefore a test with greater specificity may be preferable despite the loss of sensitivity.
As mentioned previously, a morning serum cortisol after LDDST of >5 μg/dL has a high specificity (83–100%) but a relatively low sensitivity (44–58%). In contrast, a morning serum cortisol >1.8 μg/dL after LDDST has a high sensitivity (75–100%) but a lower specificity (67–72%). Given the low specificity, there is a risk of false-positive values and overdiagnosis when using the cutoff of 1.8 μg/dL.
The use of midnight salivary cortisol levels has also been evaluated to diagnose adrenal mild hypercortisolism. However, its utility seems limited. One study that compared the MSC concentrations from patients with adrenal adenomas and known mild adrenal hypercortisolism to those with adrenal adenomas without hypercortisolism found that there was no difference. Other similar studies have replicated this outcome.
A 24 hour UFC level may also be elevated; mild increases of up to two times the upper limit of normal may support the diagnosis. However, many patients with mild hypercortisolism have normal UFC levels. As with the use of midnight salivary cortisol, mildly elevated 24 h UFC levels can help support the diagnosis of mild adrenal hypercortisolism, but normal levels do not rule out the disease.
Sequelae
Morbidity/Mortality in General
The presence of hypercortisolism, from any cause, induces a constellation of complications that ultimately impact life expectancy (see Fig. 10.1). The increase in mortality associated with hypercortisolism persists even after biochemical remission. A retrospective review of the entire population of Denmark from 1980 to 2010 found that of a cohort of 343 cases of mixed adrenal pituitary hypercortisolism, 74 patients died. The risk of death was double that of age-matched subjects with similar clinical characteristics. This effect was most pronounced 1 year after diagnosis, where the hazard ratio for death jumped to 5.2 (95% CI 2.7–9.7) when compared to the population without Cushing’s syndrome. The rates were identical when adjusted for an adrenal or pituitary cause [40].
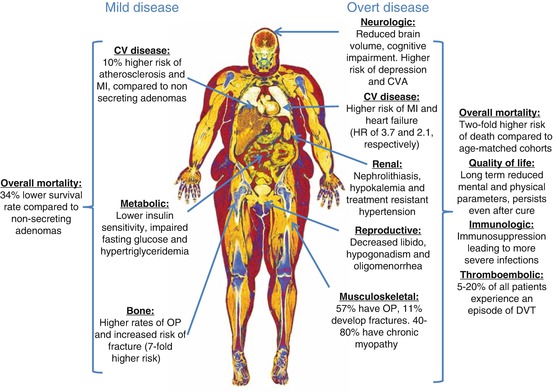
Fig. 10.1
Clinical consequences of mild and overt hypercortisolism. MI myocardial infarction, CV cardiovascular, OP osteoporosis, CVA cerebrovascular accident, HR hazard ratio, DVT deep vein thrombosis
After treatment and induction of remission, death due to cardiovascular causes continued to be higher in patients with adrenal hypercortisolism. In a cohort from New Zealand, long-term follow-up showed a HR of 1.6 (95% CI 1.3–2.1). This is striking, given that their case series revealed high biochemical cure rates of >90% with surgery. In this same retrospective analysis, the most commonly reported causes of death after exclusion of ACC were 19% from ischemic heart disease, 17% from stroke, 17% from sepsis (including pneumonia), and 11% from pulmonary embolism [41].
The duration of cortisol exposure appears to correlate with the deleterious long-term effects on morbidity and mortality even after biochemical cure. An analysis performed by Lambert and Geer et al. of 346 patients after curative treatment for Cushing’s disease showed a positive correlation between the duration of glucocorticoid exposure and death (p = 0.038) [42]. As it is difficult to ascertain the duration and degree of cortisol excess, particularly in patients with mild hypercortisolism, earlier intervention would be desirable.
The damages inflicted by adrenal Cushing’s syndrome are widespread and exert a significant impact in quality of life (see Fig. 10.1). The most commonly reported comorbidities are cardiovascular disease (CVD), venous thromboembolism (VTE), cerebrovascular accidents (CVA), infections secondary to immunosuppression, neuropsychiatric disorders, and musculoskeletal diseases [43, 44]. Similar to the mortality outcomes, the risk for the development or deterioration of these conditions persists even after the resolution of hypercortisolism through surgical means [45, 46].
Taken together, it is no surprise that adrenal hypercortisolism leads to long-term impaired quality of life. Data from a 10-year registry of 123 patients with Cushing’s syndrome showed that all mental and physical parameters of quality of life remain impaired, even after curative treatment, albeit with moderate improvements in the physical markers of disability [47].
Cardiovascular Disease
Under physiologic conditi ons, cortisol regulates metabolism and energy expenditure. Excessive cortisol secretion leads to a series of metabolic derangements, which ultimately cause significant impact on the cardiovascular system [48]. Glucocorticoids, by direct and indirect measures, promote changes in lipid and glucose utilization in myocytes and adipocytes. These changes lead to increased insulin resistance, adipocyte proliferation, and lipogenesis, which are prominent features of the metabolic syndrome [49, 50].
Hypertension , an almost universal feature of adrenal hypercortisolism, is an important contributor to the risk of cardiovascular disease. Initially, patients with CS may only have blunting of the physiologic dip in nocturnal blood pressure [51]; however, a recent study demonstrated that long-standing hypercortisolism may lead to treatment-resistant hypertension that may persist even after adrenalectomy [52]. The main mechanism underlying hypercortisolism-induced hypertension involves activation of the mineralocorticoid receptor by cortisol. Under physiologic conditions, cortisol is inactivated to cortisone in the kidney by 11-beta-hydroxysteroid-dehydrogenase 2 (11β-HSD 2). However, in the presence of very high cortisol levels, the ability of 11β-HSD 2 to inactivate to the metabolite is overwhelmed, leading to cortisol activation of mineralocorticoid receptors in the kidney, with resultant sodium reabsorption and potassium excretion.
The convergence of hypertension, impaired glucose metabolism, and dyslipidemia leads to pro-inflammatory changes in the endothelium, which results in widespread atherosclerotic buildup and cardiovascular disease [53]. Other cardiovascular effects from chronic hypertension and hypercortisolism include left ventricular hypertrophy and myocardial fibrosis, which in turn may lead to heart failure [54]. Most patients with adrenal hypercortisolism share a particular phenotype of the metabolic syndrome, featuring abdominal obesity and visceral fat accumulation. Visceral fat can be very metabolically active, overproducing adipokines (as well as other pro-inflammatory cytokines), which further increases the risk of the metabolic syndrome [55].
As discussed previously, cardiovascular disease is the main cause of the increased mortality seen with overt adrenal hypercortisolism . Acute myocardial infarction, cerebrovascular accidents, and heart failure are all much more common with overt Cushing’s syndrome (HR 3.7 95% CI 2.4–5.5, HR 2.0 95% CI 1.3–3.2, and HR 6.0 95% CI 2.1–17.1, respectively) [40]. Retrospective data from iatrogenic hypercortisolism reveals that the occurrence of cardiovascular complications exhibits a dose-dependent association with the amount of glucocorticoids prescribed and the duration of exposure. Patients with continuous use >6 months and who received a total glucocorticoid dose ≥7.5 mg of prednisolone were found to have an absolute risk difference of 6.9 more cardiovascular events per 1000 person-years, when compared to unexposed individuals. The magnitude of effect was greater with increasing steroid doses [56].
This dose-response effect seen with exogenous glucocorticoids supports the concept that mild adrenal hypercortisolism produces cardiovascular disease, as even small amounts of endogenous excess cortisol over time will likely lead to similar effects on the endothelium.
Recent epidemiological data supports this argument. In a retrospective analysis of 198 patients with AIs followed over 15 years by Di Dalmazi et al., mild hypercortisolism was found to be associated with an increased risk of cardiovascular disease. They categorized the patients as stable nonsecreting (cortisol level <1.8 μg/dL after LDDST; n = 114), stable intermediate (cortisol 1.8–5 μg/dL after LDDST), or subclinical Cushing’s (cortisol level >5 μg/dL after LDDST; n = 61). In addition, 23 patients had worsening of their hypercortisolism over the course of the study. The reported incidence of cardiovascular events was higher in the patients with stable intermediate or subclinical Cushing’s (16.7%; p = 0.04) and also with worsened secreting patterns (29.4%; p = 0.02) compared to those with stable nonsecreting adenomas (6.7%) [13].
Androulakis and colleagues recently published a case-control study which also demonstrated an association between mild hypercortisolism and increased cardiovascular risk. There were three groups: cortisol-secreting AIs (CSAIs), nonfunctioning AIs (NFAIs), and healthy controls. All patients with AIs had no known cardiovascular risk factors (hypertension, hyperlipidemia, or diabetes). CSAIs were defined as a morning cortisol after LDDST +2 standard deviations greater than the control group, which translated to a cortisol level >1.09 μg/dL, a value lower than previously suggested cutoffs. The authors measured intimal media thickness (IMT) and flow-m ediated vasodilatation (FMD), two measures associated with atherosclerosis and cardiovascular risk. The IMT was greater and FMD less in the subjects with CSAIs compared with NFAIs [57].
DeBono and colleagues have also found an association between mild hypercortisolism and an increase in cardiovascular disease and mortality. In a retrospective study of patients with adrenal adenomas, patients were categorized by their cortisol levels after LDDST. Cortisol levels of >1.8 μg/dL after LDDST were associated with decreased survival, and 50% of the deaths in that group were from cardiovascular causes [58].
As mentioned previously, the most common definition of a normal cortisol suppression following a DST is <1.8 μg/dL. However, a ROC analysis performed by Morelli et al. in search of the ideal cortisol suppression level to predict a cardiovascular event found the ideal threshold to range between 1.5 and 2.0 μg/dL [14].
Musculoskeletal Disorders
Adrenal hypercortisolism affects both the muscle and bone. This not only leads to the typical phenotype of reduced muscle volume and strength but significantly impacts quality of life and balance and may ultimately lead to an increased risk of falls and osteoporotic fractures [41, 46].
Hypercortisolism may cause myocyte damage in both an acute and chronic presentation. Acute myopathy, although uncommon, can be life threatening, as it leads to widespread and rapid involvement of the proximal, distal, and even respiratory muscles [59]. Chronic myopathy involves proximal muscles primarily, causing predominant weakness in the muscles of the pelvic girdle. This presentation is quite prevalent among patients with Cushing’s syndrome, with rates reported between 40 and 80% [60]. The main mechanisms of muscular damage are by impairment of protein synthesis through decreased amino acid uptake, inhibition of myogenesis by downregulation of myogenin, and stimulation of proteolysis through the ubiquitin-proteasome system. In the end, hypercortisolism leads to the transformation of muscle fibers from fast to slow type and muscle atrophy [46].
At the level of the bone, glucocorticoid excess leads to a multifactorial insult that can cause loss of bone density and fragility fractures [61]. Hypercortisolism leads to impairments in osteoblastogenesis as well as apoptosis of osteoblasts and osteocytes. This causes a reduction in osteoblast function (primarily th rough inhibition of the Wnt β-catenin pathway) and a major impairment of trabecular bone formation, particularly in the vertebra. At the level of the osteocyte, even small doses of exogenous glucocorticoids have been found to induce autophagy; at higher doses and more prolonged exposure, they lead to apoptosis and disruption of bone turnover that is irreversible [61–63]. Aside from the effect on bone formation, cortisol excess also causes increased bone resorption by promoting osteoclastogenesis and upregulating colony-stimulating factors and the receptor activator of nuclear factor kappa-B ligand (RANK-L) [61].
The cumulative lifetime prevalence of low impact skeletal fractures among patients with overt hypercortisolism ranges between 11 and 76% of all patients [64]. Bone impairment is more prevalent among patients with adrenal disease than with other causes of overt Cushing’s syndrome, with up to 64–100% showing some degree of bone disease. Although the most common manifestations are asymptomatic osteopenia (40–78%) and osteoporosis (22–57%), vertebral fractures can frequently be the initial finding in cases of undiagnosed CS [65, 66]. The reason for this excess risk of bone injury in patients with adrenal hypercortisolism, as opposed to ACTH-mediated causes, may be from the suppression of ACTH production from the pituitary. In a study by Zaidi et al., ACTH was found to lead to VEGF-mediated increments in osteoblast survival. Additionally, necropsy evaluation of rabbits exposed to depot methylprednisolone found that concomitant treatment with a cosyntropin infusion leads to a reduction in femoral trabecular osteonecrosis, a crucial step behind glucocorticoid bone damage [67].
The risk of osteoporosis from mild hypercortisolism has been documented in various studies. In 2009, Chiodini et al. published a multicenter, retrospective study done in Italy evaluating the bone mineral density (BMD), prevalence of vertebral fractures, and bone quality in patients with AIs with and without subclinical hypercortisolism (SH). SH in this study was defined as two out of three of the following: UFC >70 μg/24 h, morning cortisol >3 μg/dL after LDDST, and ACTH <10 pg/mL. The primary endpoint measured was the spinal deformity index (SDI), in which the number and severity of vertebral fractures are integrated. The SDI has been associated with vertebral fracture risk over time. The study showed that the patients with AIs (SH+) had lower BMD at the lumbar spine (trabecular bone) and femoral neck (cortical bone), more metabolic syndrome, and increased spinal deformity index compared to those with AIs (SH-) and controls (no AI). The odds ratio for vertebral fractures was 7.27 (p = 0.0001) for patients who were SH+ regardless of age, MD, menopause status, and gender.
More recently in 2011, the same group conducted a multicenter, prospective longitudinal study evaluating SDI and vertebral fractures at baseline and after 12 and 24 months in patients who were SH+ and SH− with AIs. The results were similar to those of the retrospective study. The authors reported that the prevalence of fractures and SDI was higher in the SH+ group regardless of age, sex, BMI, MBD, and menopausal status. Those who were SH+ had a higher risk of vertebral fractures than those who were SH− with an odds ratio of 12.264 (p = 0.001) [68].
Immunosuppression
Adrenal hypercortisolism directly impacts the innate immune system , both by affecting its humoral and cellular responses. The humoral immune system is primarily affected through downregulation of pro-inflammatory cytokines and blunted lymphocyte proliferation. The adaptive component of the immune response is also impaired, as evidence has shown glucocorticoid excess also leads to inhibition of maturation and proliferation of antigen-presenting cells and T-helper lymphocyte subclasses 1 and 2 (mediators of the cellular and humoral immune response, respectively). These effects lead to decreases in the production of IgG and IgE immunoglobulins, which decreases the ability of the immune system to recognize and ultimately opsonize and kill pathogens [69]. The cellular immune response is impaired both by the previously mentioned selective downregulation of Th1 lymphocytes and by directly impairing the action, maturation, and proliferation of neutrophils, monocytes, eosinophils, and macrophages [70]. The final result is an increase in susceptibility toward intracellular pathogens and opportunistic infections [71–73].
Although immunosuppression is the major consequence of glucocorticoid excess during the active phase of hypercortisolism, an increased susceptibility to autoimmunity has also been documented after curative treatment. This phenomenon is poorly understood, although it may involve a rebound in the immune response after immunosuppression, as well as a selective Th1/Th2 lymphocyte imbalance [43]. The degree of immunosuppression is determined both by the length and severity of hypercortisolism. A systematic review of eight cohorts of patients with Cushing’s syndrome showed a HR of 2.4 (95% CI 1.0–5.9) for the risk of infection, 3 years before diagnosis; the risk peaked in the immediate postoperative period, with a HR of 38.2 (95% CI 16.9–86.1). Therefore, infections are important determinants of perioperative death [40].
The most commonly reported Gram-positive infections are those caused by Staphylococcus, Streptococcus, and Listeria pathogens. The most commonly reported Gram-negative infections are those from members of the Enterobacteria and Legionella family of organisms. Community-acquired and nosocomial infections in hypercortisolemic patients tend to present in more severe and invasive forms. Fungal (e.g., Candida and Aspergillus) and viral (e.g., herpes zoster, CMV, and influenza) infections also tend to be more severe and common [69, 71].
Thromboembolic Disease
The pro-thrombotic effect of hypercortisolism is twofold, both by inducing the activation of the coagulation pathway and by inhibition of fibrinolysis [74]. Prior research has shown that glucocorticoids mainly induce the activation of coagulation factors VIII and IX and vWF. Evidence of increased production of fibrinogen has also been found with hypercortisolism. Fibrinogen plays an important role both for the coagulation pathway and with primary hemostasis through mediation of platelet aggregation. Endogenous hypercortisolism may additionally influence other components of hemostasis, by acting through the upregulation of fibrinolysis and subsequent increased thrombin generation as well as by directly stimulating thrombocytosis and platelet function [75].
The effects from the many metabolic dysregulations with glucocorticoid excess also play a role in thrombosis. Factors such as hypertension, hyperhomocysteinemia, and hyperglycemia with subsequent hyperinsulinemia and dyslipidemia indirectly influence hemostasis, by upregulating vWF and tissue plasminogen activato sr inhibitor 1/tPa concentrations and activating the extrinsic coagulation pathway [74]. A meta-analysis of 476 cases of pituitary and adrenal hypercortisolism showed a high rate of thrombotic events (1.9–2.5%) before surgery; postoperatively, the rate climbed to as high as 5.6% with a maximum incidence of 20% of subjects in one study [76].
Other Complications
Under physiologic conditions, cortisol exerts significant control in the regulation of many neuropsychiatric, reproductive, and dermatologic functions. Although epidemiologic data for overt hypercortisolism has clearly shown the impact of excess glucocorticoids in these systems, the influence of mild hypercortisolism on these organs has not yet been elucidated.
The effects of glucocorticoid excess on the nervous system are not completely understood. Deleterious events affecting neuroplasticity, the secretion of stimulatory neurotransmitters such as glutamate, and suppression of neurogenesis in the hippocampus have been postulated as causal factors [77]. The psychiatric disorders in patients with endogenous hypercortisolism can be quite debilitating and resistant to even curative treatment [40, 77]. The most common manifestation is a psychoactive/hypomanic form of major depression (50–81%), although anxiety (66%) and bipolar (30%) disorders are quite frequent as well [78]. Long-standing hypercortisolism, if left untreated, may ultimately lead to the loss of brain volume and neurocognitive impairment [79].
Adrenal hypercortisolism negatively affects both the reproductive and sexual health of men and women [2, 5]. Excess cortisol can block gonadotropin hormone release by reducing the amplitude and frequency of gonadotropin-releasing hormone production in the hypothalamus, while visceral adiposity may lead to abnormalities in the metabolism of sex steroids and sex hormone-binding globulin as well as androgen excess [80]. Hypercortisolism may also cause direct damage to the ovaries and testis, leading to reduced primordial follicles with cortical stromal fibrosis and Leydig cell impairment with tubular atrophy, respectively [80, 81]. These phenomena disrupt the hypothalamic-pituitary axis and ultimately may lead to hypogonadism and infertility [82]. Decreased libido, hypogonadism in men, and menstrual irregularities in women are common (24–90%, 50–70%, and 43–80%, respectively) [40]. Long-standing hypogonadism may also negatively impact bone health and increase the risk of osteoporosis, particularly in men [64].
Related posts:
 Pheochromocytomas and Paragangliomas: Genetics and Pathophysiology
Pheochromocytomas and Paragangliomas: Genetics and Pathophysiology
 Adrenal Zonation and Development
Adrenal Zonation and Development
 Diagnosis and Management of Adrenal Insufficiency
Pheochromocytomas and Paragangliomas: Genetics and Pathophysiology
Diagnosis and Management of Adrenal Insufficiency
Pheochromocytomas and Paragangliomas: Genetics and Pathophysiology
 Genetics and Pathophysiology of Congenital Adrenal Hyperplasia
Genetics and Pathophysiology of Congenital Adrenal Hyperplasia
 Diagnosis and Management of Adrenal Insufficiency
Diagnosis and Management of Adrenal Insufficiency
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


