Fig. 14.1
Schematic representation of adoptive T cell transfer therapy using T cells genetically modified with chemokine receptor. Tumor mass is excised from the patient and TILs (tumor-infiltrating lymphocytes) are isolated from the tumor. TILs are transduced with the chemokine receptor matching with the ligand abundantly produced by tumor cells. Chemokine receptor positive T cells are expanded in cell culture using medium enriched with IL-2. Expanded modified T cells are infused back into patient to have better homing potential and effective tumor cell killing
The published data regarding overexpression of chemokine receptors on T cells directing antitumor effector T cells to tumor sites are scarce. It was found, for example, that CCL2 and CCR4 play a role in T-cell chemoattraction by melanoma in vitro [160] and that tumor infiltration of T cells is strongly associated with high CXCL9 and CXCL10 expression in melanoma in in situ hybridization studies [161]. CXCL12 is shown to enhance T-cell migration toward melanoma in vitro [162], but also cause chemorepulsion in other systems [163]. The selective expression of chemokine receptors by different subsets of T cells can determine specific trafficking of these subsets to tissues expressing the appropriate chemokine. Thus, for example, CCR7, expressed by naïve T cells, facilitates migration to lymph nodes where the ligands for this receptor, CCL21 and CCL19, are produced [164]. The expression of chemokine receptors by T cells and chemokines at sites of antigenic challenge determine the specific traffic of lymphocytes. For example, the ligands for CXCR3, CXCL10, and CXCL9 [165], which can be expressed by activated monocytes, fibroblasts, keratinocytes, and endothelial cells [166], may enable cells bearing CXCR3 to traffic preferentially to IFN-γ producing inflammatory sites. Though, the complete T-cell/tumor chemotactic network is still to be explored, as well as the pattern of chemokine receptors on clinically derived ex vivo cultured T cells. Our understanding of how to exploit chemotactic signals in order to manipulate reactive T cells to better reach tumor sites is far from being complete.
Tumor-reactive T cells do not necessarily express the appropriate receptor for chemokines produced at the site of tumors, as discussed earlier. For example, CXCL1 is produced by a large percentage of melanomas [167], but its receptor, CXCR2, is expressed only in a small subset of T cells [168]. In a study to identify which chemokines are produced by cancer cells and which chemokine receptors are expressed by cultured T cell, CXCL1 and CCL5 were identified in a series of human tumor cell lines and fine needle aspirates; in addition, it was determined that several chemokine receptors are expressed by cultured human T cells, including CCR1, CCR2, CCR4, CCR5, CXCR3, and CXCR4. Activated lymphocytes may also be a source of chemokines; in a strategy to direct T cells toward chemokines expressed by tumors, CXCL1 was chosen because it was produced by tumors but not by T cells themselves. The absence of CXCL1 by T cells may be an important requisite for trafficking to tumors because endogenous chemokine production may block or cause downregulation of chemokine receptor on T cells. However, T cells did not express the receptor CXCR2, and therefore, T cells were transduced with a retroviral vector encoding CXCR2. T cells expressing CXCR2 were responsive in vitro toward both recombinant protein and tumor-derived chemokine. Furthermore, it was demonstrated that CXCL1 was able to induce the secretion of the proinflammatory cytokine IFN-γ by transduced T cells, thereby extending the possibility of antitumor functions in modified T cells. This study demonstrates the feasibility of redirecting the migration properties of T cells toward chemokines secreted by tumors [159].
Several approaches have been applied to find out the mechanism of unsuccessful migration and homing of effector T cells into tumor microenvironment. Methods such as Affymetrix gene expression profiling on a series of metastatic melanoma biopsies were performed to reveal T-cell-associated transcripts that could be of potential use. The presence of lymphocytes also correlates with the expression of defined chemokine genes. In this approach, a subset of six chemokines (CCL2, CCL3, CCL4, CCL5, CXCL9, and CXCL10) was confirmed by protein array and quantitative reverse transcription PCR to be preferentially expressed in tumors that contained T cells. Corresponding chemokine receptors were found to be upregulated on human CD8+ effector T cells, and transwell migration assays confirmed the ability of each of these chemokines to promote migration of CD8+ effector cells in vitro. Screening by chemokine protein array identified a subset of melanoma cell lines that produced a similar broad array of chemokines. These melanoma cells more effectively recruited human CD8+ effector T cells when implanted as xenografts in nonobese diabetic/severe combined immunodeficient (NOD/SCID) mice in vivo. Chemokine blockade with specific antibodies inhibited migration of CD8+ T cells. This study suggests that lack of critical chemokines in a subset of melanoma metastases may limit the migration of activated T cells, which in turn could limit the effectiveness of antitumor immunity [169]. The majority of tumors, including neuroblastoma, produce the chemokine CCL2. In one recent study, it has been shown that forced co-expression of chemokine receptor CCR2b, along with chimeric antigen receptor specific for the tumor-associated antigen GD2, enhanced the tumor trafficking of activated T cells [170]. As a result, adoptively transferred T cells co-modified with both CCR2b and GD2-CAR had greater antitumor activity in vivo. To better understand the importance of homing of the adoptively transferred T cells to all tumor sites in sufficient number, a similar study was done exploiting endogenous chemotactic signals in order to manipulate and enhance the directional trafficking of transferred T cells toward melanoma. Based on chemokine profiling of 15 melanoma cultures, it was shown that CXCL1 and CXCL8 are abundantly expressed and secreted from melanoma cultures. However, the complementary analysis on 40 melanoma patient-derived tumor-infiltrating lymphocytes (TILs) proves that the corresponding chemokine receptors are either not expressed (CXCR2) or expressed at low levels (CXCR1). Using the in vitro transwell system, it was demonstrated that tumor-infiltrating lymphocytes (TILs) preferentially migrate toward melanoma and that endogenously expressing CXCR1 TIL cells are significantly enriched among the migrating lymphocytes. The role of the chemokine receptor CXCR1 is validated by the enhanced migration of CXCR1 engineered TIL cells toward melanoma or recombinant CXCL8. Cytotoxicity and interferon secretion activity are unaltered by CXCR1 expression profile. Taken together, these results mark CXCR1 as a candidate for genetic manipulations to enhance the trafficking of adoptively transferred T cells [171]. This approach is complementary and potentially synergistic with other genetic strategies designed to enhance antitumor potency. In a similar study, the introduction of chemokine receptor CXCR2 gene into tumor-specific T cells was shown to have enhanced localization to tumors and improved antitumor responses against melanoma expressing chemokine CXCL1 and CXCL8 [80]. The chemokine CXCL16 also plays an important role in T-cell-mediated antitumor immune responses: mice lacking CXCR6, the receptor for CXCL16, displayed reduced recruitment of activated effector T cells in breast tumor tissue and impaired tumor regression [139]. A similar study was done to suggest that the capacity of adoptively transferred T cells to home to tumors may be, in part, dictated by the species and amounts of tumor-derived chemokines, in particular CCL2 [172].
The chemokine CCL2 is highly secreted by malignant pleural mesotheliomas, but the corresponding chemokine receptor, CCR2, is minimally expressed on activated human T cells genetically transduced with a chimeric antibody receptor (CAR) directed to the tumor antigen mesothelin (mesoCAR T cells). The chemokine receptor CCR2b was thus transduced into mesoCAR T cells using a lentiviral vector and the modified T cells were used to treat established mesothelin-expressing tumors. CCR2b transduction led to CCL2-induced calcium flux and increased transmigration, as well as augmentation of in vitro T-cell killing ability. A single intravenous injection of 20 million mesoCAR CCR2b T cells into immunodeficient mice bearing large, established tumors (without any adjunct therapy) resulted in a 12.5-fold increase in T-cell tumor infiltration by day 5 compared with mesoCAR T cells. This was associated with significantly increased antitumor activity. This study concluded that CAR T cells bearing a functional chemokine receptor can overcome the inadequate tumor localization that limits conventional CAR targeting strategies and can significantly improve antitumor efficacy in vivo [173]. Identifying the mechanisms of trafficking as well as suitable chemokine and chemokine receptor pair that can enhance the tumor trafficking and migration of adoptively transferred T cells with antitumor effect is therefore of the utmost importance in optimizing therapeutic benefits.
In one of the most recent studies, the introduction of chemokine and receptor axis CCL2/CCR2 is shown to potentiate in vivo anti-lung cancer reactivity mediated by CD8 +ve T cells [174]. WT1 is a well-known tumor antigen expressed to various degrees by human lung cancer cells and the small cell lung cancer cell line used as a target which produces high amount of chemokine CCL2. Lymphocytes were engineered to co-express both WT1-specific TCR and chemokine receptor CCR2 not only via CCL2-tropic tumor trafficking but also via CCL2-enhanced WT1-responsiveness. Based on this observation, the clinical feasibility of this strategy for adoptive immunotherapy against human lung cancer can be addressed in the future.
One potentially interesting chemokine is CX3CL1 or Fractalkine, having an important role in leukocyte migration. Neuroblastoma cells overexpressing Fractalkine are capable of inducing migration, adhesion, and IFN-γ secretion by immune effector cells [134]. The role of this chemokine/receptor pair CX3CL1/CX3CR1 has been well established in glioblastoma multiforme, an aggressive tumor of the central nervous system, and in the adenocarcinoma of the pancreas [175, 176] and is now being investigated extensively in colorectal cancer. Recent studies by our group show the overexpression of Fractalkine in colorectal cancer assessed in human clinical samples [177]. Fractalkine/ CX3CL1 is a proinflammatory chemokine that chemoattracts and activates CX3CR1+ leukocytes such as CD8+, CD4+, and γδ T lymphocytes, natural killer (NK) cells, dendritic cells (DCs), and monocytes. Leukocyte trafficking is modulated by multiple signal transduction pathways including CX3CL1-CX3CR1 signaling [178]. High expression of CX3CL1 was positively correlated with good prognosis and the number of TIL in colorectal carcinoma [135]. High expression of CX3CL1 by tumor cells correlates with a good prognosis and increased tumor-infiltrating CD8+ T cells, NK cells, and DCs in breast carcinoma [179]. The choice of the chemokine receptor CX3CR1 to enhance the homing potential of adoptively transferred T cells is currently being studied in mouse tumor models.
14.9 Concluding Remarks
Several strategies were introduced to enhance the efficacy of ACT [180]. The development of targeted small molecules, mAbs, and biological therapies that demonstrate greater efficacy and lower toxicity remains highly desirable in hematology and oncology in general. In the context of biological therapies, T lymphocyte-based treatments have enormous potential. Over the past decade, it has become clear that the adoptive transfer of ex vivo expanded antigen-specific cytotoxic T lymphocytes promotes sustained antitumor effects in patients. Because of this compelling clinical evidence and the concomitant development of methodologies for robust gene transfer to human T lymphocytes, the field has rapidly evolved, offering new opportunities to extend T-cell-based therapies [181]. To exert a therapeutic effect, adoptively transferred tumor-specific cytotoxic T lymphocytes must traffic to sites of tumor burden, exit the circulation, and infiltrate the tumor microenvironment. This can be addressed with the idea that chemokines play a major role in antitumor immune responses. As such, they hold great potential in cancer immunotherapy for increasing immune cell infiltration of the tumor microenvironment to facilitate productive immune interactions. Studies in experimental tumor models and cancer patients clearly demonstrate the potential of chemokine immunotherapy and suggest that future trials should seek to incorporate chemokines into therapy protocols. The possibility of developing novel strategies aimed at improving T-cell homing to tumors used alone or in combination with current regime of adoptive T-cell therapies against cancer, such as introduction of antigenic receptor or signaling molecules, may prove to be more efficient and holds great promises in several other oncological settings (Fig. 14.2).
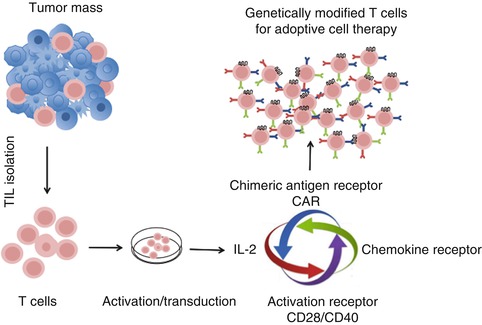
Fig. 14.2
Optimizing adoptive T cell transfer therapy using T cells genetically modified with multiple factors. Tumor mass is excised from the patient and TILs (tumor-infiltrating lymphocytes) are isolated from the tumor. TILs are transduced with the chemokine receptor matching with the ligand abundantly produced by tumor cells along with CAR specific to the antigen expressed by tumor cells as well as with enhanced T cell activation signal genes such as CD28 or CD40. Modified T cells are expanded in cell culture using medium enriched with IL-2. Expanded modified multifunctional T cells are accessed further for their antitumor activity in ACT
Acknowledgment
This work was funded by the FP7 European People Programme ATTRACT. I.S. was a recipient of a Marie Curie fellowship from FP7 ITN Number 238778.
References
1.
Siegel R, DeSantis C, Virgo K, Stein K, Mariotto A, Smith T, et al. Cancer treatment and survivorship statistics. CA Cancer J Clin. 2012;62:220–41.PubMed
3.
Rosenberg SA, Yang JC, Restifo NP. Cancer immunotherapy: moving beyond current vaccines. Nat Med. 2004;10:909–15.PubMedCentralPubMed
4.
Galluzzi L, Vacchelli E, Eggermont A, Fridman WH, Galon J, Sautès-Fridman C, et al. Trial watch: adoptive cell transfer immunotherapy. Oncoimmunology. 2012;1:306–15.PubMedCentralPubMed
5.
Blattman JN, Greenberg PD. Cancer immunotherapy: a treatment for the masses. Science. 2004;305:200–5.PubMed
6.
Rosenberg SAS. Cell transfer immunotherapy for metastatic solid cancer–what clinicians need to know. Nat Rev Clin Oncol. 2011;8:577–85.PubMed
7.
Smith FOF, Downey SGS, Klapper JAJ, Yang JC, Sherry RM, Royal RE, et al. Treatment of metastatic melanoma using interleukin-2 alone or in conjunction with vaccines. Clin Cancer Res. 2008;14:5610–8.PubMedCentralPubMed
8.
Atkins MB, Hsu J, Lee S, Cohen GI, Flaherty LE, Sosman JA, et al. Phase III trial comparing concurrent biochemotherapy with cisplatin, vinblastine, dacarbazine, interleukin-2, and interferon alfa-2b with cisplatin, vinblastine, and dacarbazine alone in patients with metastatic malignant melanoma (E3695): a trial coordinated by the Eastern Cooperative Oncology Group. J Clin Oncol. 2008;26:5748–54.PubMedCentralPubMed
9.
Hodi FS, O’Day SJ, McDermott DF, Weber RW, Sosman JA, Haanen JB, et al. Improved survival with ipilimumab in patients with metastatic melanoma. N Engl J Med. 2010;363:711–23.PubMedCentralPubMed
10.
Robert C, Thomas L, Bondarenko I, O’Day S, Weber J, Garbe C, et al. Ipilimumab plus dacarbazine for previously untreated metastatic melanoma. N Engl J Med. 2011;364:2517–26.PubMed
11.
Rosenberg SA, Lotze MT, Muul LM, Leitman S, Chang AE, Ettinghausen SE, et al. Observations on the systemic administration of autologous lymphokine-activated killer cells and recombinant interleukin-2 to patients with metastatic cancer. N Engl J Med. 1985;313:1485–92.PubMed
12.
Galluzzi LL, Senovilla LL, Zitvogel LL, Kroemer GG. The secret ally: immunostimulation by anticancer drugs. Nat Rev Drug Discov. 2012;11:215–33.PubMed
13.
Tartour E, Pere H, Maillere B, Terme M, Merillon N, Taieb J, et al. Angiogenesis and immunity: a bidirectional link potentially relevant for the monitoring of antiangiogenic therapy and the development of novel therapeutic combination with immunotherapy. Cancer Metastasis Rev. 2011;30:83–95.PubMed
14.
Finn OJO. Cancer vaccines: between the idea and the reality. Nat Rev Immunol. 2003;3:630–41.PubMed
15.
Sasada T, Noguchi M, Yamada A, Itoh K. Personalized peptide vaccination: a novel immunotherapeutic approach for advanced cancer. Hum Vaccin Immunother. 2012;8(9):1309–13.PubMedCentralPubMed
16.
Frankenberger B, Schendel DJ. Third generation dendritic cell vaccines for tumor immunotherapy. Eur J Cell Biol. 2012;91:53–8.PubMed
17.
Kantoff PW, Higano CS, Shore ND, Berger R, Small EJ, Penson DF, et al. Sipuleucel-T immunotherapy for castration-resistant prostate cancer. N Engl J Med. 2010;363:411–22.PubMed
18.
Small EJ, Schellhammer PF, Higano CS, Redfern CH, Nemunaitis JJ, Valone FH, et al. Placebo-controlled phase III trial of immunologic therapy with sipuleucel-T (APC8015) in patients with metastatic, asymptomatic hormone refractory prostate cancer. J Clin Oncol. 2006;24:3089–94.PubMed
19.
Beer TM, Bernstein GT, Corman JM, Glode LM, Hall SJ, Poll WL. Randomized trial of autologous cellular immunotherapy with sipuleucel-T in androgen-dependent prostate cancer. Clin Cancer Res. 2011;17:4558–67.PubMed
20.
Kantoff PW, Schuetz TJ, Blumenstein BA, Glode LM, Bilhartz DL, Wyand M, et al. Overall survival analysis of a phase II randomized controlled trial of a poxviral-based PSA-targeted immunotherapy in metastatic castration-resistant prostate cancer. J Clin Oncol. 2010;28:1099–105.PubMedCentralPubMed
21.
Abastado J-P. The next challenge in cancer immunotherapy: controlling T-cell traffic to the tumor. Cancer Res. 2012;72:2159–61.PubMed
22.
Rosenberg SAS, Yang JCJ, Sherry RMR, Kammula US, Hughes MS, Phan GQ, et al. Durable complete responses in heavily pretreated patients with metastatic melanoma using T-cell transfer immunotherapy. Clin Cancer Res. 2011;17:4550–7.PubMedCentralPubMed
23.
Gattinoni LL, Powell DJD, Rosenberg SAS, Restifo NPN. Adoptive immunotherapy for cancer: building on success. Nat Rev Immunol. 2006;6:383–93.PubMedCentralPubMed
25.
Tacken PJ, de Vries IJM, Torensma R, Figdor CG. Dendritic-cell immunotherapy: from ex vivo loading to in vivo targeting. Nat Rev Immunol. 2007;7:790–802.PubMed
26.
Barnes DW, Loutit JF. Treatment of murine leukaemia with x-rays and homologous bone marrow. II. Br J Haematol. 1957;3:241–52.PubMed
27.
Bortin MM, Rimm AA, Saltzstein EC. Graft versus leukemia: quantification of adoptive immunotherapy in murine leukemia. Science. 1973;179:811–3.PubMed
28.
Bortin MM, Truitt RL, Rimm AA, Bach FH. Graft-versus-leukaemia reactivity induced by alloimmunisation without augmentation of graft-versus-host reactivity. Nature. 1979;281:490–1.PubMed
29.
Mathé G, Amiel JL, Schwarzenberg L, Cattan A, Schneider M. Adoptive immunotherapy of acute leukemia: experimental and clinical results. Cancer Res. 1965;25:1525–31.PubMed
30.
Schwarzenberg LL, Mathé GG, Schneider MM, Amiel JLJ, Cattan AA, Schlumberger JRJ. Attempted adoptive immunotherapy of acute leukaemia by leucocyte transfusions. Lancet. 1966;2:365–8.PubMed
31.
Horowitz MMM, Gale RPR, Sondel PMP, Goldman JMJ, Kersey JJ, Kolb HJH, et al. Graft-versus-leukemia reactions after bone marrow transplantation. Blood. 1990;75:555–62.PubMed
32.
Weiden PL, Sullivan KM, Flournoy N, Storb R, Thomas ED. Antileukemic effect of chronic graft-versus-host disease. N Engl J Med. 1981;304:1529–33.PubMed
33.
Collins RHR, Rogers ZRZ, Bennett MM, Kumar VV, Nikein AA, Fay JWJ. Hematologic relapse of chronic myelogenous leukemia following allogeneic bone marrow transplantation: apparent graft-versus-leukemia effect following abrupt discontinuation of immunosuppression. Bone Marrow Transplant. 1992;10:391–5.PubMed
34.
Goldman JM, Gale RP, Horowitz MM, Biggs JC, Champlin RE, Gluckman E, Hoffmann RG, Jacobsen SJ, Marmont AM, McGlave PB. Bone marrow transplantation for chronic myelogenous leukemia in chronic phase. Increased risk for relapse associated with T-cell depletion. Ann Intern Med. 1988;108:806–14.PubMed
35.
Slavin SS, Naparstek EE, Nagler AA, Ackerstein AA, Kapelushnik JJ, Or RR. Allogeneic cell therapy for relapsed leukemia after bone marrow transplantation with donor peripheral blood lymphocytes. Exp Hematol. 1995;23:1553–62.PubMed
36.
Kolb HJH, Mittermüller JJ, Clemm CC, Holler EE, Ledderose GG, Brehm GG. Donor leukocyte transfusions for treatment of recurrent chronic myelogenous leukemia in marrow transplant patients. Blood. 1990;76:2462–5.PubMed
37.
Alexandros Spyridonidis ML. A long road of T-cells to cure cancer: from adoptive immunotherapy with unspecific cellular products to donor lymphocyte infusions and transfer of engineered tumor-specific T-cells. Am J Blood Res. 2012;2:98–104.PubMedCentralPubMed
39.
Related posts:
 Novel Strategy of Cancer Immunotherapy: Spiraling Up
Novel Strategy of Cancer Immunotherapy: Spiraling Up
 Dendritic Cell Vaccines for Cancer Therapy: Fundamentals and Clinical Trials
Psychoneuroendocrinoimmunotherapy of Cancer
Dendritic Cell Vaccines for Cancer Therapy: Fundamentals and Clinical Trials
Psychoneuroendocrinoimmunotherapy of Cancer
 Polarization of Tumor Milieu: Therapeutic Implications
Polarization of Tumor Milieu: Therapeutic Implications
 Photodynamic Therapy and Antitumor Immune Response
Photodynamic Therapy and Antitumor Immune Response
 Role of γδ T Lymphocytes in Cancer Immunosurveillance and Immunotherapy
Role of γδ T Lymphocytes in Cancer Immunosurveillance and Immunotherapy
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


