Fig. 5.1
Lean adipose in insulin-sensitive state. The adipocyte is responsive to insulin stimulation, thus prompting glucose uptake via Glut4 transporter, and free fatty acid uptake (FFA). Glucose is converted to glycerol and is combined with FFA to form triglycerides. PPARγ promotes triglyceride synthesis and lipoprotein lipase activity. As noted, the drug class of thiazolidinediones (TZD) increases PPARγ activity. Adapted from Guilherme et al. [60]
More recently, the endocrine role of adipocytes has been gaining attention given its relative complexity and underlying pathologic involvement in a number of disease states. Adipose tissue synthesizes and secretes a number of different proteins with systemic action, termed adipocytokines, or adipokines [38–40]. While more than 100 different adipokines have been identified, proteomic studies have indicated the possibility of several hundreds. Their roles vary, and include controlling appetite, insulin sensitivity, blood pressure, hemostasis, and inflammation [12, 41, 42]. They also affect several organs, including the liver, pancreas, and muscle, along with the central nervous system [43]. The adipocyte’s role in inflammation has been of particular interest given its ability to secrete a variety of the well-known cytokines and chemokines including TNF-α, IL-1β, IL-6, IL-10, and several others [44, 45]. Few others have garnered particular interest as well, notably leptin and adiponectin. Leptin, first identified by Friedman and colleagues in 1994, serves a primarily antidiabetic role modulating food intake and energy expenditure, regulating hepatic lipogenesis, and enhancing muscle fatty acid oxidation [43, 46–48]. It has been shown to protect mice from obesity as well [49]. Thus, leptin concentration increases as the proportion of stored fat increases [50]. Adiponectin has roles in insulin sensitizing, as an anti-inflammatory agent, and is anti-atherogenic in character [51–53].
5.3 Obesity and Changes to the Adipocyte and WAT
Globally, it has long been observed that the prevalence of obesity has been on the rise. This is not only true in the adult population, but also alarmingly so in the pediatric population, with potentially significant impact on the future of health care [54]. Long-known associated risks of obesity include type 2 diabetes mellitus (T2DM), cardiovascular disease, arthritis, and increased mortality, among others [55–58]. These pathologic outcomes are the product of significant changes resulting primarily from an energy imbalance, and start at the level of cellular mechanisms involving the adipocyte, its relation its neighboring cells, and beyond with its interplay with the body as a whole.
With persistent consumption of calories in excess of expenditure naturally comes the demand for increasing storage capacity. During states of excess, lipogenic enzymes, localized in the cytoplasm and endoplasmic reticulum (ER), synthesize triglyceride, which is then incorporated into the fat droplet. Adipocytes have a significant capacity to synthesize and store triglycerides. Early on, adipocytes compensate for the increase FFA load by increased expression of enzymes associated with triglyceride synthesis [59]. With progression, accommodation occurs via hypertrophy and hyperplasia [60]. Regional tissue variability associated with adipogenesis has been observed. Intraperitoneal (visceral) fat general enlarges via hypertrophy, whereas regions of subcutaneous fat tend to expand via hyperplasia [61]. It has been suggested in animal models that hyperplasia occurs first in increasing the number of preadipocytes, and then proceeding to mature adipocytes [62]. While much remains to be delineated, larger cells release more FFA, which may underlie the significance of fat distribution and elevated free fatty acid levels in obesity. This was portrayed in a mouse model in which overdevelopment of subcutaneous adipose tissue resulted improved glucose and lipid homeostasis [63]. Thus, and not surprisingly, visceral adipose tissue is significantly linked to increased risk of cardiovascular disease and a strong predictor for developing T2DM, and may act as a surrogate marker for ectopic fat distribution, namely the liver and muscle [64, 65]. Another regional difference is the significantly greater FFA release in upper body in addition to the aforementioned visceral fat, when compared to the nonobese or lower body-obese state. Hence, lower body stores, mainly the gluteo-femoral region, may be viewed as a protective metabolic region [66]. Aging and sedentary lifestyles also serve as factors in increasing the ratio of visceral to subcutaneous fat [67].
Histologically, beyond the changes to the adipocytes themselves, macrophage infiltration increases in WAT. Macrophages typically organize in a ring around the adipocyte; such organization is specific to adipose tissue, and more prevalent in visceral WAT than subcutaneous WAT, and intimates their role in the phagocytosis of necrotic adipocytes [68]. In contrast to the relative balance of M1 and M2 macrophages, these macrophages are M1, and thus pro-inflammatory in nature. T-cell infiltration is also present in WAT without an increase in systemic circulation, presumably due to dysfunctional adipokine release, discussed below [69]. Not surprisingly, accompanying the pro-inflammatory state is fibrosis of the extracellular matrix, organized in clusters and fibrotic bundles, and surrounding adipocytes [70]. Interestingly, M2 macrophages expressing higher levels of tumor growth factor β (TGFβ), which stimulates collage VI production, were found in greater number. They also express increased IL-1, suggesting more of a pro-inflammatory role contrasting M2 macrophages in the non-obese state [71]. It has been shown that patients with a higher degree of adipose tissue fibrosis were found to lose less fat mass after gastric bypass, and that fibrosis may serve a protective role in omental WAT in limiting hypertrophy and its associated deleterious effects [70].
5.4 Excessive FFA, Ectopic Fat Deposition, and Insulin Resistance
Naturally, with progression of obesity comes an increased release of FFAs into the blood stream [72]. Circulating levels of FFAs are a significant mediator connecting obesity with insulin resistance. Elevated levels have been shown to cause insulin resistance in both animals and humans, with an acute decrease in levels resulting in enhanced insulin activity and peripheral glucose uptake [73, 74]. With the accumulation of fatty acids and its metabolites, activation via phosphorylation of serine kinases such as JNK and IKK results in blocking and inactivating insulin receptors. Said mechanism is present in a multitude of cells including adipocytes, myocytes, and hepatocytes [75–77]. Knockout mouse models of JNK and IKK show resistance to the effects of high fat diet on insulin receptor signaling [78, 79]. JNK is required for FFA-mediated macrophage release of inflammatory cytokines such as TNFα, IL-6, and MCP-1 [80]. Additionally, FFAs may induce insulin resistance via their activation of Toll-like receptors (TLR) on adipocytes and macrophages, as mutation of TLR4 prevents obesity and insulin resistance in mice on a high fat diet [60, 81, 82]. Mice with myeloid-specific TLR4 deletion became obese on a high fat diet but were protected from insulin resistance [83]. Cells from TLR4 knock out mice were unresponsive to the inflammatory effects of FFAs [82, 84]. One of the end results is decreased membrane mediated glucose transport via disruption of Glut4. As such, hyperinsulinemia ensues as compensation [84] (Fig. 5.2).
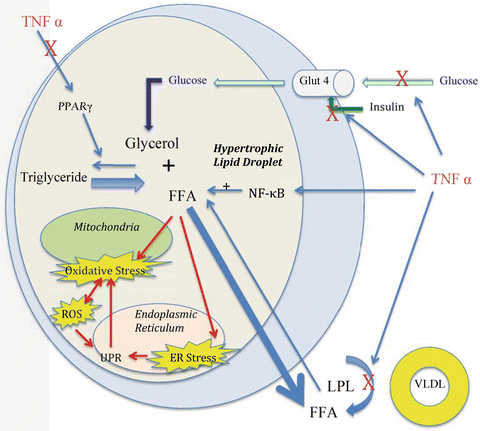
Fig. 5.2
Hypertrophic adipocyte in inflammatory state. Adipocyte hypertrophy results in increased free fatty acid (FFA). With the increased FFA comes mitochondrial oxidative stress and endoplasmic reticulum stress. This in turn results in increased reactive oxidative species (ROS) and activation of the unfolded protein response (UPR). Inflammatory cytokines like TNFα lead to increased activation of the proinflammatory pathway NF-κB, decreased cellular insulin responsiveness, and decreased PPARγ and lipoprotein lipase activity. Adapted from Guilherme et al. and de Ferranti et al. [60, 101]
At a cellular level, obesity decreases the rate of lipid turnover, and is related to decreased catecholamine stimulated lipolysis given the sympathetic innervation of WAT [85–87]. The primary mediators of lipolysis are adipose triglyceride lipase (ATGL), hormone-sensitive lipase (HSL), and monoglyceride lipase (MGL) [88]. HSL is responsible for converting triacylglycerol to diacylglycerol and monoacylglycerol, while ATGL participates in fat mobilization and MGL in the final hydrolysis of the 2-monoacylglycerols produced by HSL [89]. ATGL is important for basal lipolysis, whereas HSL is important during catecholamine-stimulated lipolysis, via the SNS, as previously noted [90]. Obesity results in significantly decreased HSL and ATGL in obese patients. Regionally, ATGL is not significantly different between omental and subcutaneous storage depots, but HSL does differ and is much higher in omental stores correlating with adipocyte size and fasting plasma insulin concentrations [91]. The activity of HSL is further affected by a blunted catecholamine response seen in obesity, correlating with the notion that catecholamines exert their strongest influence over visceral fat [92]. Hypertrophy, observed more so in visceral obesity, correlates with a decrease in lipolytic activity governed by a higher density of α-2 adrenergic receptors, and a lower density of lipolytic β-1/2 adrenergic receptors presumably in an effort to limit contributing to the already elevated circulating FFA levels [92, 93].
Once FFA storage capacity has been met coupled with the decreased lipid mobilization, a spillover effect is observed, at which point organs are exposed to the deleterious effects of unoxidized FFA. Increased hepatic FFA uptake results in hepatic steatosis, then worsening insulin resistance and hyperglycemia in addition to leading to nonalcoholic steatotic hepatitis (NASH) [94]. Evidence suggests that hepatic fat is strongly associated with insulin resistance [95, 96]. As visceral fat increases, so does hepatic delivery via the splanchnic bed, more selectively so than increases in subcutaneous fat do [94]. This in turn stimulates hepatic VLDL-triglyceride production [97]. FFA deposition and intracellular accumulation may also be observed in muscle, pancreatic β-cells, and the heart, which exacerbate insulin resistance perpetuating a vicious cycle [98]. Elevated circulating FFA is also associated with inhibition of carbohydrate oxidation and glycogen synthesis in muscle [99]. The direct lipotoxicity to pancreatic β-cells is significant as it can lead to their dysfunction and apoptosis hindering their capacity to accommodate the metabolic derangement at a time of increased insulin requirements [100, 101]. In rodents, lipid accumulation in cardiac myocytes results in cellular damage and ventricular dysfunction [98]. The effects of ectopic distribution of adipose tissue are observed as well in lipodystrophic patients with defects in triglyceride storage in adipose tissue, and in mice without WAT, as both populations exhibit severe insulin resistance. Upon surgical transplantation of functional adipose tissue in mice, there is a dramatic reversal of hyperglycemia, hyperinsulinemia, and insulin resistance [102, 103].
As noted earlier, the effective nature of thiazolidinediones is due to their action on PPARγ receptors which stimulate FFA uptake by subcutaneous adipocytes resulting in decreased ectopic fat distribution and the increased insulin sensitivity [104]. PPARγ is also present in macrophages where they negatively regulate a multitude of inflammatory genes [105]. In PPARγ knockout mice, insulin resistance is impaired, and worsens following high-fat feeding [106, 107]. An important aspect of adipocyte dysfunction arises from downregulation of PPARγ by inflammatory cytokines, and in particular TNFα, both from macrophages and adipocytes. TNFα has been shown to negatively impact PPARγ in many ways, including transcription, posttranscription, and translation [108]. When treated with TNFα, PPARγ mRNA is more rapidly turned over in adipocytes [109]. Another factor negatively affecting PPARγ expression in preadipocytes and adipocytes is hypoxia. This may also be the underlying reason for the inhibited adipocyte differentiation in a hypoxic state [110].
5.5 Hypoxia and Inflammation
As indicated by the histologic changes accompanying obesity, inflammatory changes are a significant driver of pathogenicity as well. With the advent of hyperplasia and more so hypertrophy comes macrophage infiltration and aggregation around necrotic adipocytes. Adipocytes enlarge to accommodate for the increased FFA load. However, their growth will then reach a limit given restraints from oxygen tension, which could explain the ensuing cell death and initiation of macrophage infiltration [68, 111]. The degree of infiltration correlates with obesity and insulin resistance regardless of BMI. Thus, of two similarly obese patients, the patient with increased macrophage infiltration will exhibit worse insulin resistance [112]. The concept of hypoxia-induced inflammation is supported given that adipocytes can increase in size to up to 200 μM in the obese state which is similar to or greater than that of normal oxygen diffusion distance, and although lean patients have the ability to increase postprandial blood flow to WAT, no such increase in blood flow is observed in obese patients [113, 114]. Both qualitative and quantitative studies via the hypoxyprobe system and needle-type fiber-optic O2 sensor, respectively, have also demonstrated hypoxia present in adipose tissue in the obese mouse model [8, 115, 116]. Macrophage tissue infiltration is evident in hypoxic tissue areas as well, thus providing a link between hypoxia, adipocyte stress and apoptosis, and inflammation [114]. In humans, the PO2 of oxygen has been observed to be decreased in the adipose tissue of obese patients, when compared to lean counterparts, with PO2 levels inversely correlating with percent body fat [117].
One of the main aspects lending support to hypoxia as a factor in inflammation is the up-regulation of hypoxic induced factors, mainly hypoxia-inducible factor 1α (HIF-1α) , a key regulator of oxygen homeostasis [118]. Using a transgenic mouse model of HIF-1α overexpression, adipose tissue fibrosis and increased local inflammation are observed [119]. With selective pharmacologic and genetic inhibition of HIF-1α activity, high-fat diet-fed obese mice demonstrated significant metabolic improvements and reduced inflammation in WAT [120]. A key role in the increased inflammation may be the role HIF-1α has in downregulating the expression of PPARγ [110].
5.6 Inflammation, Endoplasmic Reticulum, and Mitochondria
As the demand for increased lipid storage expands, so does the capacity and activity of the adipocyte endoplasmic reticulum, which is responsible for synthesizing proteins, forming lipid droplets, and regulating cholesterol [101]. Thus, with obesity and increasing FFA load, ER “stress” develops. This state is characterized by its functional disturbance in which case proper folding and modification of proteins and lipid droplet creation are disturbed [101]. The ER is able to identify the imbalance in supply and production via the Unfolded Protein Response (UPR), which is subsequently activated through its three arms: PKR-like eukaryotic initiation factor 2α kinase (PERK), inositol-requiring enzyme-1 (IRE-1), and activating transcription factor-6 (ATF-6) [121, 122]. PERK activation leads to decreased protein translation and increased expression of a multitude of genes, including those related to apoptosis [123]. Another UPR response is to induce transcription of chaperones to assist with the increasing volume of unfolded proteins. IRE-1 contributes to the increase in chaperone proteins produced to assist with the unfolded protein load, while ATF-6 is responsible for increasing the expression of ER degradation-enhancing α-mannosidase like protein (EDEM) facilitating the clearance of chaperone proteins [124, 125]. The increased chaperone load is likely responsible for the increased oxidative stress via increased reactive oxidative species (ROS) from mediating oxidation-reduction reactions [121, 126]. IRE-1 also upregulates JNK and IKK resulting in increased expression of inflammatory genes responsible for increased cytokine production [127, 128]. While the goal of such changes brought about by UPR are for preserving cell function and stressor accommodation, the end result of inadequate adaptation may yet be apoptosis [121].
Mitochondria also exhibit signs of distress, not only in adipocytes, but in multiple organs as well. Increases in FFA causes increased release of ROS in obese patients [129]. Lipid infusion in lean human subjects results in decreased mRNAs for many mitochondrial genes [130]. Mitochondrial dysfunction is evident in the pancreas, liver, and muscle as well [125, 131, 132]. In the pancreas, insulin production is negatively affected by ROS. In muscle, there is decreased fat oxidation and ectopic fat accumulation contributing to insulin resistance [133]. Increased intramyocellular lipid content has been observed with down-regulation of genes encoding mitochondrial respiratory complexes I–IV, and genes responsible for cytochrome c oxidase complexes I and III which are subunits of the electron transport chain [134, 135]. PPARγ is responsible for controlling mitochondrial gene subsets, and with its reduced activity may contribute to the decreased mitochondrial function [136]. Also contributing to the decreased mitochondrial function is the increased amount of inflammatory cytokines [137]. Notably, with the mitochondrial dysfunction comes decreased fatty acid oxidation and metabolites that inhibit glucose transport [138].
The presence of ROS associated with obesity is thought to play a central role in the decreased mitochondrial activity [139]. Once again, with the elevated FFA levels in obesity comes increased ROS [140]. In diabetic patients, endothelial cells portray elevated ROS via NADPH oxidase activation [141]. Mice overexpressing superoxide dismutase 2 have decreased levels of ROS, improved hepatic insulin sensitivity, and normalization of glucose and insulin levels [142]. In rats, soleus muscle exposure to nitric oxide donors caused decreased insulin sensitivity, and were associated with decreased insulin-stimulated phosphorylation of insulin receptor (IR) and insulin receptor substrate-1 (IRS-1), critical in the insulin intracellular signaling pathway [143]. Other kinases are also activated, including JNK and NFκB, further inhibiting IRS-1 progressing insulin resistance [144–146].
Uncoupling proteins (UCP) are mitochondrial inner membrane proteins that mediate the coupling of electrons through the electron transport chain, primarily allowing for a proton leak through the inner membrane [147]. UCP2 is expressed in several tissues, and because of its distribution in multiple tissues, it has been hypothesized to have a significant role in decreasing ROS, thus protecting against oxidative stress [148, 149]. At the same time, several studies have shown that increased UCP2 production leads to decreased insulin secretion from pancreatic β cells, predisposing to diabetes mellitus [150–152]. In UCP2 knockout mice, pancreatic islets have increased insulin secretion in response to glucose when compared to wild-type mice [153]. Furthermore, double-mutant leptin/UCP2 knockouts also have improved beta cell function independent of obesity [153]. FFAs seem to be a key mediator of UCP2 as in preadipocytes, UCP2 mRNA expression increases significantly when exposed to FFAs [154].
5.7 Inflammation and Adipocytokines
The complex role of the adipocyte as an endocrine organ has gained significant attention given its ability to secrete several different types of factors. With the infiltration of macrophages into adipose tissue, cytokine secretion accompanies and influences the adipose tissue environment. FFAs have been shown to strongly stimulate TNF-α production in macrophages via TLR4 receptor activating NFκB [155]. Further activation from ER stress and UPR along with secretion from adipocytes also increases local TNF-α concentration [121]. Conversely, TNF-α secretion inhibits lipoprotein lipase activity, thus increasing FFA release from adipocytes [156]. Thus, a vicious paracrine loop develops that perpetuates the macrophage-adipocyte inflammatory state [157]. TNF-α leads to activation of JNK1 via phosphorylation of IRS-1 and its inhibition, as mentioned above, linking TNF-α with insulin resistance [158]. The cycle is worsened as adipocyte hypertrophy develops given their capacity for increased FFA release [63]. TNF-α also decreases adiponectin secretion, whose actions result in increased insulin sensitivity by decreasing hepatic glucose production and increasing fatty acid oxidation in both liver and muscle [159]. Multiple studies have implicated low adiponectin levels as a strong indicator for the development of insulin resistance and T2DM [160, 161]. Adiponectin-deficient mice develop insulin resistance in the setting of elevated TNF-α and reduced responsiveness to PPARγ [161]. Adiponectin acts via its two receptors, AdipoR1 and AdipoR2. AdipoR1 is universally expressed whereas AdipoR2 is primarily localized to the liver. Mouse knockouts of these two receptors have increased lipid accumulation, and inflammation, and exhibit increased insulin resistance [43, 160, 162].
Whereas adiponectin production is decreased in hypertrophic and inflamed tissue, leptin production is significantly increased [50]. In leptin-deficient mice models and humans, leptin administration leads to decreased hyperphagia and reduced body mass [163]. However, it has also been seen to increase IL-6 and TNF-α production by macrophages [164]. Leptin acts via a number of different pathways including the JAK-STAT pathway which regulates the expression of anorexic neuropeptides, and the phosphatylinositol-3-kinase pathway which stimulates insulin sensitivity in peripheral tissues [43, 165]. The interesting concept of leptin resistance, similar to insulin resistance, has also been proposed, and been shown in states of inflammation whereby subsequent metabolic stress negatively regulates leptin signaling [166]. Similar resistance has been proposed to be evident in the hypothalamus as well [167]. Overall, energy expenditure and appetite remains poorly controlled even as leptin levels increase in obese patients [43, 163].
5.8 Conclusion
The complexity that characterizes insulin resistance is underscored by the remarkable evolution of our understanding of the adipocyte and its role in metabolic homeostasis. The mechanisms underlying the progression from an insulin sensitive state to that of adipocyte dysfunction, inflammation, and local and systemic insulin resistance are complex, and include a series of vicious cycles that perpetuate the inflammatory state. As we continue to delineate the mechanisms that accompany the changes in the adipocyte correlating with obesity, potential therapeutic targets will continue to emerge. For now, surgery will continue to serve as one of the mainstays in the treatment of obesity, and will remain a source for potential answers in reversing some of the deleterious effects of obesity and insulin resistance.
References
1.
Cannon B, Hedin A, Nedergaard J. Exclusive occurrence of thermogenin antigen in brown adipose tissue. FEBS Lett. 1982;150:129–32.PubMed
2.
Cannon B, Nedergaard J. Brown adipose tissue: function and physiological significance. Physiol Rev. 2004;84:277–359.PubMed
3.
Cinti S, Zancanaro C, Sbarbati A, Cicolini M, Vogel P, Ricquier D, Fakan S. Immunoelectron microscopical identification of the uncoupling protein in brown adipose tissue mitochondria. Biol Cell. 1989;67:359–62.PubMed
4.
Frontini A, Rousset S, Cassard-Doulcier AM, Zingaretti C, Ricquier D, Cinti S. Thymus uncoupling protein 1 is exclusive to typical brown adipocytes and is not found in thymocytes. J Histochem Cytochem. 2007;55:183–9.PubMed
5.
Cinti S. The adipose organ: morphological perspectives of adipose tissues. Proc Nutr Soc. 2001;60:319–28.PubMed
6.
Cinti S. Transdifferentiation properties of adipocytes in the adipose organ. Am J Physiol Endocrinol Metab. 2009;297(5):E977–86. doi:10.1152/ajpendo.00183.2009.PubMed
7.
Hausman GJ. Anatomical and enzyme histochemical differentiation of adipose tissue. Int J Obes. 1985;9 Suppl 1:1–6.PubMed
8.
Trayhurn P. Hypoxia and adipocyte physiology: implications for adipose tissue dysfunction in obesity. Annu Rev Nutr. 2014;34:207–36. doi:10.1146/annurev-nutr-071812-161156.PubMed
9.
Weisberg SP, McCann D, Desai M, Rosenbaum M, Leibel RL, Ferrante Jr AW. Obesity is associated with macrophage accumulation in adipose tissue. J Clin Invest. 2003;112(12):1796–808.PubMedCentralPubMed
10.
Xu H, Barnes GT, Yang Q, Tan G, Yang D, Chou CJ, Sole J, Nichols A, Ross JS, Tartaglia LA, Chen H. Chronic inflammation in fat plays a crucial role in the development of obesity-related insulin resistance. J Clin Invest. 2003;112(12):1821–30.PubMedCentralPubMed
11.
Aron-Wisnewsky J, Tordjman J, Poitou C, Darakhshan F, Hugol D, Basdevant A, Aissat A, Guerre-Millo M, Clément K. Human adipose tissue macrophages: m1 and m2 cell surface markers in subcutaneous and omental depots and after weight loss. J Clin Endocrinol Metab. 2009;94(11):4619–23. doi:10.1210/jc.2009-0925.PubMed
12.
Trayhurn P. Hypoxia and adipose tissue dysfunction in obesity. Physiol Rev. 2013;93(1):1–21. doi:10.1152/physrev.00017.2012.PubMed
13.
Bastard J, Feve B. Physiology and physiopathology of adipose tissue. Paris: Springer; 2013.
14.
Tchkonia T, Thomou T, Zhu Y, Karagiannides I, Pothoulakis C, Jensen MD, Kirkland JL. Mechanisms and metabolic implications of regional differences among fat depots. Cell Metab. 2013;17(5):644–56. doi:10.1016/j.cmet.2013.03.008.PubMedCentralPubMed
15.
Shuster A, Patlas M, Pinthus JH, Mourtzakis M. The clinical importance of visceral adiposity: a critical review of methods for visceral adipose tissue analysis. Br J Radiol. 2012;85(1009):1–10. doi:10.1259/bjr/38447238.PubMedCentralPubMed
16.
Kahn SE, Prigeon RL, Schwartz RS, Fujimoto WY, Knopp RH, Brunzell JD, Porte Jr D. Obesity, body fat distribution, insulin sensitivity and Islet beta-cell function as explanations for metabolic diversity. J Nutr. 2001;131(2):354S–60.PubMed
17.
Peinado JR, Jimenez-Gomez Y, Pulido MR, Ortega-Bellido M, Diaz-Lopez C, Padillo FJ, Lopez-Miranda J, Vazquez-Martínez R, Malagón MM. Cellular and molecular basis of functional differences among fat depots. Proteomics. 2010;10(18):3356–66. doi:10.1002/pmic.201000350.PubMed
18.
Tchoukalova YD, Votruba SB, Tchkonia T, Giorgadze N, Kirkland JL, Jensen MD. Regional differences in cellular mechanisms of adipose tissue gain with overfeeding. Proc Natl Acad Sci U S A. 2010;107(42):18226–31. doi:10.1073/pnas.1005259107.PubMedCentralPubMed
19.
Tchkonia T, Morbeck DE, Von Zglinicki T, Van Deursen J, Lustgarten J, Scrable H, Khosla S, Jensen MD, Kirkland JL. Fat tissue, aging, and cellular senescence. Aging Cell. 2010;9(5):667–84. doi:10.1111/j.1474-9726.2010.00608.x.PubMedCentralPubMed
20.
Youngstrom TG, Bartness TJ. Catecholaminergic innervation of white adipose tissue in the Siberian hamster. Am J Physiol. 1995;268(3 Pt 2):R744–51.PubMed
21.
Bartness TJ, Liu Y, Shrestha YB, Ryu V. Neural innervation of white adipose tissue and the control of lipolysis. Front Neuroendocrinol. 2014;35:473. doi:10.1016/j.yfrne.2014.04.001. pii: S0091-3022(14)00043-0.PubMedCentralPubMed
22.
Mansfeld G, Muller F. Der Einfluss der Nervensystem auf die Mobilisierung von Fett. Arch Physiol. 1913;152:61–7.
23.
Hales CN, Luzio JP, Siddle K. Hormonal control of adipose tissue lipolysis. Biochem Soc Symp. 1978;43:97–135.PubMed
Related posts:
 Effect of Bariatric Surgery on Insulin Secretion
Understanding Diabetes Mellitus
Neural Modulation in the Treatment of Obesity
Bile Acids, the Microbiome and Metabolic Disease-Implications for Surgery
Effect of Bariatric Surgery on Insulin Secretion
Understanding Diabetes Mellitus
Neural Modulation in the Treatment of Obesity
Bile Acids, the Microbiome and Metabolic Disease-Implications for Surgery
 Understanding Diabetes Mellitus: Pathophysiology
Understanding Diabetes Mellitus: Pathophysiology
 Medical Approaches to Weight-Centric Management of Obese Patients with Type 2 Diabetes
Medical Approaches to Weight-Centric Management of Obese Patients with Type 2 Diabetes
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


