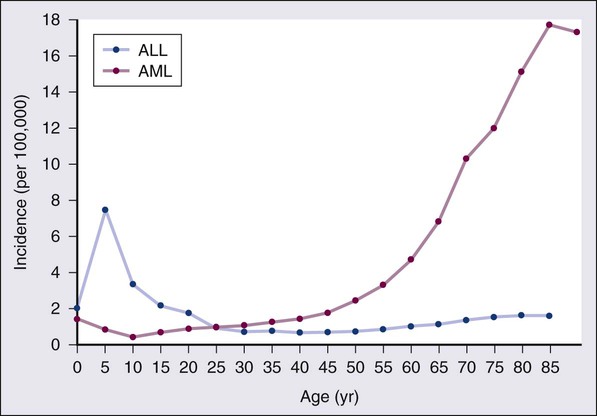
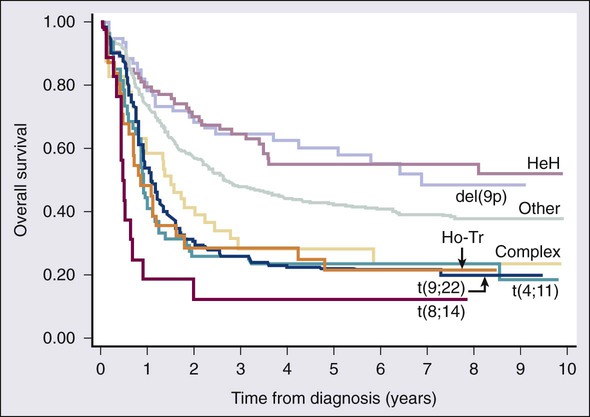
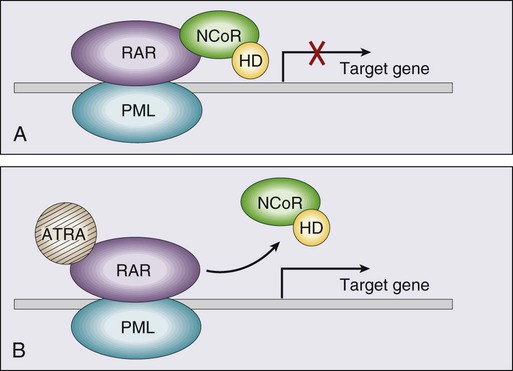
• There were 13,780 new cases of acute myeloid leukemia (AML) and 6,050 new cases of acute lymphocytic leukemia (ALL) in the United States in 2012, resulting in 10,200 deaths from AML and 1440 from ALL. The incidence of AML is relatively low until age 35 and then rises almost exponentially, whereas ALL peaks in incidence in young children. Risk factors for the development of acute leukemia include exposure to benzene, ionizing radiation, or previous chemotherapy. • The acute leukemias are clonal disorders, with all leukemic cells in a given patients descending from a common precursor. Recent genomewide analyses of leukemia have demonstrated considerable complexity with a number of recurrent potential “driver” mutations and a much larger number of random “passenger” mutations in individual cases. • The diagnosis of acute leukemia is generally made by bone marrow examination. In the past, the classification of acute leukemia depended heavily on morphologic examination of the leukemic cells. This is no longer the case, and currently the most important elements of classification are the immunophenotype (to distinguish AML from ALL), cytogenetics, and mutational analyses. In AML, three primary-risk groups are recognized. Favorable-risk patients are those with CBF translocations, and those with normal cytogenetics and either NMP1 or CEBPA mutations without mutations in FLT3-ITD. Unfavorable-risk patients are those with abnormalities of 3q, 5 or 7, and those with complex cytogenetics. The intermediate-risk group includes all those not classified as favorable or unfavorable. Acute promyelocytic leukemia, characterized by t(15;17), is a separate entity and requires specific therapy. In ALL, the favorable-risk group includes those with high hyperdiploidy, and del9q. The unfavorable-risk group includes t(4;11), low hypodiploidy/near triploidy, and those with complex cytogenetics. All others comprise the intermediate-risk group. Patients with t(9;22) (Ph+ ALL) and mature B-cell ALL including Burkitt leukemia comprise separate categories of ALL that require specific treatment. Patients With AML Who Are Candidates for Intensive Therapy • Induction chemotherapy that includes an anthracycline plus cytarabine will result in a complete remission in approximately 70% of patients. Postremission therapy depends on the risk group. Favorable-risk patients are generally treated with three or four cycles of consolidation chemotherapy, including high-dose cytarabine. With such treatment, approximately 50% of patients will be cured. Patients with intermediate-risk disease should undergo allogeneic transplantation while in first remission if they have a matched sibling or matched unrelated donor. The use of partially matched donors in this setting is less agreed on. Allogeneic transplantation using matched siblings, matched unrelated donors, or partially matched cord blood is recommended for patients with unfavorable-risk disease in first remission. Patients With AML Who Are Not Candidates for Intensive Therapy • Low-dose cytarabine is a generally accepted therapy for patients with AML who are not candidates for intensive therapy. Recent alternatives include decitabine, azacitidine, or clofarabine. • Induction therapy should include all-trans-retinoic acid (ATRA) alone with an anthracycline and, for higher-risk patients, cytarabine. The inclusion of arsenic trioxide during consolidation therapy appears to prolong survival. Patients With ALL Who Are Candidates for Intensive Therapy • Induction therapy in ALL includes a combination of vincristine, prednisone, an anthracycline, and asparaginase, with cyclophosphamide sometimes included. With such regimens, 75% to 90% of patients will achieve a complete remission. Postremission therapy generally involves six to eight courses of intensive consolidation therapy, several of which contain high-dose methotrexate, cytarabine, and asparaginase, and several of which include the same drugs used for initial remission induction. Some form of central nervous system (CNS) prophylaxis is required, as is low-dose maintenance therapy. High-risk patients should receive an allogeneic transplant in first remission if at all possible. The role of allogeneic transplantation for patients with standard-risk disease is more controversial. • A tyrosine kinase inhibitor (imatinib, nilotinib, or dasatinib) should be included as part of induction and consolidation therapy. Allogeneic transplantation is recommended in first remission if an appropriate donor is available. • Therapy should include, along with the usual drugs used for remission induction, high doses of fractionated cyclophosphamide, high-dose methotrexate and cytarabine, and rituximab. According to the American Cancer Society, there will be 13,780 new cases of AML and 6050 cases of ALL in the United States in 2012, resulting in 10,200 deaths from AML and 1440 from ALL.1 The incidence of AML has a bimodal pattern with a modest peak among infants, a decline in childhood, and then an exponential rise with advancing age (Figure 98-1). ALL likewise has a bimodal pattern, but the initial peak occurs among children aged 1 to 4, with a decline between ages 20 and 60, and a more modest rise in advanced years.2 The rates for all forms of acute leukemia are higher in males than females, and tend to be higher in non-Hispanic whites, with the exception of higher rates of acute promyelocytic leukemia (APL) and B-cell ALL among Hispanic whites.2 The concordance rate of acute leukemia in identical twins is virtually 100% if one twin develops leukemia during the first year of life, but then declines with age. “Pure familial leukemia” is a term used to describe those rare syndromes caused by a single mutation that leads to leukemia without other manifestations; to date, three have been described involving RUNX1, CEBPA, and GATA2.3,4 The incidence of acute leukemia is also increased in primary bone marrow failure syndromes, such as Diamond-Blackfan, Shwachman-Diamond, and dyskeratosis congenita, as well as in syndromes involving defective DNA repair, such as Fanconi anemia and Bloom syndrome. Acute leukemia is also increased in some of the syndromes associated with loss of tumor suppressor gene function such as Li-Fraumeni syndrome, and in syndromes caused by abnormal chromosomal numbers, including 21+ and XXY.3 Aside from these rare syndromes, there does not appear to be any familial aggregation of acute leukemia; that is, first-degree relatives of patients with acute leukemia are not at increased risk for developing the disease.5 Human T cell lymphotropic virus type 1 (HTLV1) is an enveloped, single-strand RNA virus that is associated with the development of a distinct form of adult T cell leukemia found in southwestern Japan, the Caribbean basin, and Africa. The virus can be spread vertically from mother to fetus or horizontally by sexual contact or blood products. Although endemic in the areas noted above, the virus only causes leukemia in 2% to 4% of those infected, and has a very long latency period, estimated at 30 years or more.6 Epstein-Barr virus, a DNA herpes family virus, is associated with the endemic African form of Burkitt lymphoma/leukemia. Ionizing radiation is leukemogenic. An increased incidence of AML, ALL and chronic myelogenous leukemia (CML) was seen in survivors of the atomic bombings of Hiroshima and Nagasaki, which began approximately 1.5 years after exposure, peaked at 7 years, and returned to baseline 25 years later. The risk of leukemia was also increased in individuals treated with radiation for ankylosing spondylitis in the 1940s. Although most of the data from humans showing the leukemogenic potential of ionizing radiation come from individuals exposed to high dose rates, occupational exposure to low dose-rate radiation has more recently been associated with the development of leukemia.7 Although the exact percentage is uncertain, as much as 10% of AMLs and a smaller percentage of ALLs are thought to be the consequence of prior exposure to chemotherapy or radiation for a primary malignancy or autoimmune disorder.8 Treatment-related leukemias can be grouped into several syndromes. AML developing after alkylating agent therapy has a latency of 5 to 7 years, often first appears as a myelodysplastic syndrome (MDS), and is frequently associated with abnormalities involving the long arms of chromosomes 5 or 7. All alkylating agents are likely leukemogenic, with an increased incidence of leukemia observed after prolonged exposure, following the use of dose-intense regimens, or when used in certain combinations. Treatment-related AML is also seen following exposure to topoisomerase II inhibitors, such as etoposide, teniposide or anthracyclines. These leukemias develop relatively rapidly, often within 2 years of exposure, are not generally preceded by a myelodysplastic phase, and frequently have rearrangements involving 11q23, the locus for MLL (the mixed-lineage leukemia gene) or 21q22. A number of other drugs have also been implicated in the subsequent development of acute leukemia, including bimolane, an agent used to treat psoriasis, and recently, lenalidomide, an agent used in the treatment of myeloma or myelodysplasia.9 Patients with lymphoma who receive autologous hematopoietic cell transplants are at increased risk for the development of leukemia, which may, in part, be predicted by the therapy received before the transplant. Not all treatment-related leukemias fall into the discrete categories described here. Secondary leukemias with inv(16), t(9;22), and abnormalities involving 3q21 have been reported. Although there remains some debate, in general, patients with treatment-related leukemias do not fare as well as patients with de novo leukemia, even after adjusting for known risk factors.10 Recent genome-wide analysis of leukemia has demonstrated considerable complexity with a number of recurrent potential “driver” mutations, perhaps up to 10, and a much larger number of random “passenger” mutations in each individual case. Thus overt leukemia is not the result of a single mutation. Careful studies in both AML and ALL have further found considerable variability in the mutations among the individual leukemic cells in any single case. The pattern of variability suggests that the development of leukemia is not a simple progressive accumulation of mutations in an essentially linear direction but rather is a dynamic process shaped by multiple cycles of mutation acquisition and clonal selection.11,12 Within the clonal populations of cells in patients with acute leukemia, there must be a subpopulation of cells capable both of self-renewal and proliferation. Such cells have been termed “leukemia stem cells.” Although there is no gold standard for their identification, the ability of human leukemic cells to engraft long-term in immunodeficient NOD/SCID mice is generally accepted as a marker for a leukemia stem cell. In human AML, the subpopulation of cells capable of long-term engraftment in immunodeficient mice is within the CD34++CD38– CD 123+ fraction and comprises only a small percent (0.2 to 100/106) of the leukemic cells present.13 However, many cases of AML do not engraft even in the more permissive NOD/SCID/IL2R immunodeficient mouse, including almost all cases of acute promyelocytic leukemia (APL), and thus the ability of leukemic cells to grow in an immunodeficient mouse is not a true assay for the leukemia stem cell. AML cells are typically 12 to 20 µm in diameter with large nuclei, multiple nucleoli, and azurophilic granules in the cytoplasm. ALL cells are typically smaller than AML blasts and are devoid of granules (Figure 98-2). For more than 3 decades, the French-American-British (FAB) system was used to describe and classify morphologic subtypes of AML and ALL. Morphologic subclassification of acute leukemia adds little prognostic or therapeutic information beyond that provided by cytogenetic and mutational categorization, but still remains part of the overall World Health Organization (WHO) classification schema. Acute leukemias can be categorized according to the cell surface antigens they express as determined by their reactivity to a panel of monoclonal antibodies (Table 98-1). The cell surface antigens expressed largely reflect the level of differentiation of the leukemic cell, and this information provides some prognostic information, especially in ALL. In addition, the antigens on leukemia blasts are almost always present in abnormal combinations or concentrations, allowing for the detection of as few as 0.01% abnormal cells in an otherwise normal looking bone marrow using multidimensional flow cytometry. Table 98-1 Immunophenotype of Acute Leukemia A small percentage of cases of acute leukemia express characteristics of both lymphoid and myeloid cells, and have recently been defined by the WHO as mixed-phenotype acute leukemia (MPAL).14 To fulfill the WHO criteria, the leukemia must express the myeloid-specific marker myeloperoxidase (either by flow cytometry or cytochemistry), and either a strictly specific T lymphoid marker (cytoplasmic CD3) or a B lymphoid marker (strong expression of CD19 or weak expression of CD19 together with expression of at least three other B-cell-associated markers). Many of these patients will have rearrangements of BCR/ABL or MLL or have complex cytogenetics. The median survival for this rare group of patients appears to be short, in the range of 18 months, and they should be considered as a high-risk group when selecting appropriate therapy. For AML, the Southwest Oncology Group (SWOG) and the Medical Research Council (MRC) each developed and adopted cytogenetic risk classifications over two decades ago based on the outcomes of large prospective clinical trials.15,16 The resulting classifications, though similar, are not identical. For example, as shown in Table 98-2, in the SWOG categorization, patients with clonal abnormalities not recognized as within one of the three major groups are assigned to a fourth category with a predicted treatment outcome somewhere between the intermediate- and unfavorable-risk groups. And there are minor differences between classification schemas in the distinction between intermediate and unfavorable risk cytogenetics. More recently, in 2010, the MRC revised their classification system, with the major change being the movement of patients with various translocations involving chromosome 11 (with the exception of t(9;11) and t(11;19)) from the intermediate-risk group into the unfavorable group.17 Although these classification schemas differ slightly, they are each able to distinguish three major groups of patients. Among patients age <65 treated with standard chemotherapy, those with a favorable karyotype have complete response (CR) rates in the 85% to 90% range and a 5-year overall survival (OS) of 50% to 60%. Those with intermediate-risk cytogenetics have CR rates of 65% to 75%, and a 5-yr OS of 35% to 45%. And for those with unfavorable-risk cytogenetics, a CR rate of 45% to 55% and a 5-yr OS of 10% to 20% can be expected. Table 98-2 Cytogenetic Classification of AML The finding of a single monosomy is a relatively common finding in AML, with monosomy 7 being the most frequent. The presence of a single monosomy (excluding loss of a sex chromosome) is associated with a negative outcome, with a 12% OS at 4 years in the Dutch experience.18 Patients with two or more distinct monosomies, or a single monosomy but with an additional structural abnormality, have an even worse prognosis, with a predicted 4-year survival of less than 4%.18 Thus this category of patient (two monosomies or one plus an additional structural abnormality) has been defined as having a “monosomal karyotype” and identifies a group of patients with a very dismal prognosis if treated with conventional chemotherapy. There is obvious overlap among patients with unfavorable, complex, and monosomal karyotypes. All patients with complex karyotypes fall within the unfavorable-risk group and comprise between a third and half of the total, and almost all patients with a monosomal karyotype are within the complex category, accounting for about two-thirds of those patients. As in AML, cytogenetic analysis plays an important role in the evaluation of patients with ALL (Table 98-3). The most common abnormality in ALL, seen in 20% to 30% of cases, is t(9;22)(q34;q11), which results in the Philadelphia chromosome. Other common translocations in adult B-cell ALL include t(4;11)(q21;q23), which is seen in 7% of cases, involves the MLL and AF4 gene and is associated with a poor prognosis, and t(8;14)(q24.1;q32), which is seen in 2% to 4% of adult B-cell ALL, involves c-MYC and the immunoglobulin heavy chain, and is the translocation associated with Burkitt lymphoma. T-cell ALLs often have translocations involving chromosomes 7 or 14 at T-cell receptor enhancer gene sites. The other most common cytogenetic changes seen in adult ALL involve del(9p) seen in 5% to 9% of cases, del(6q) seen in 5% to 7% and del(13q) seen in 3% to 5%. Abnormalities in chromosome number are also common. High hyperdiploidy (51 to 65 chromosomes) is seen in as many as 10% of cases, whereas low hypodiploidy (30 to 39 chromosomes)/near triploidy (60 to 78 chromosomes) is seen in 4%. Approximately 5% will have a complex karyotype. Compared with those cases of ALL with normal cytogenetics, those with del(9q) or high hyperdiploidy appear to have a more favorable outcome, whereas those with t(4;11), t(8;14), and del(6q), low hypodiploidy/near triploidy, or complex cytogenetics do worse (Figure 98-3).19 Table 98-3 Major Cytogenetic Categories in Adult ALL Acute promyelocytic leukemia (APL), which accounts for approximately 8% of adult AML cases, is almost always associated with t(15;17)(q22;q11.2), a translocation that fuses the promyelocytic leukemia gene (PML) on chromosome 15 to the retinoic acid receptor alpha (RARa) gene on chromosome 17. The resultant PML/RARa fusion protein acts as a dominant negative inhibitor of normal PML function and of the function of RXRa, an important heterodimeric partner of RARa. PML/RARa recruits a nuclear co-repressor (NCoR) and the molecules sin3 and histone deacetylase (HD) (Figure 98-4). HD deacetylases histones, a process that in turn inhibits binding of transcription factors, thus inhibiting the expression of genes required for hematopoietic differentiation. The unique activity of all-trans-retinoic acid (ATRA) in APL appears to be explained by its ability to bind to the PML/RARa fusion, changing its configuration and releasing the attached nuclear co-repressor. This then allows subsequent transcription and gene expression with subsequent maturation of the APL blasts. A number of other translocations, including t(5;17) and t(11;17), involve RARa and result in an APL-like phenotype. These leukemias are, however, generally unresponsive to ATRA because clinically achievable concentrations of the drug do not result in release of the NCoR-HD complex. Core binding factor is a heterodimeric transcription factor made up of two subunits, CBFα (also known as RUNX-1) and CBFβ. CBF plays a role in the transcriptional activation of a number of genes required for normal hematopoietic differentiation. The t(8;21) abnormality seen in approximately 8% of adult AML cases results in the fusion of the RUNX1 gene on chromosome 21 to the MTG8 gene on chromosome 8. The RUNX1/MTG8 fusion protein acts as a dominant negative inhibitor of the wild-type RUNX-1 gene (Figure 98-5). AML characterized by t(8;21) has a favorable prognosis. Similarly, inv(16)(p13;q22) and t(16;16)(p13;q22) both result in the fusion of CBFβ at 16q22 to the smooth-muscle myosin heavy chain (MYH11) gene at 16p13. As in the case of t(8;21), the resultant fusion protein acts as a dominant negative regulator of transcription. AML cases with involvement of 16q22 account for approximately 9% of adult AML cases, have a unique myelomonocytic morphology, and like t(8;21) leukemias have a favorable prognosis.
Acute Leukemias in Adults
Epidemiology
Etiology
Genetic Predisposition
Viruses
Radiation
Prior Therapy
Pathobiology
Clonality and Clonal Architecture
The Leukemic Stem Cell
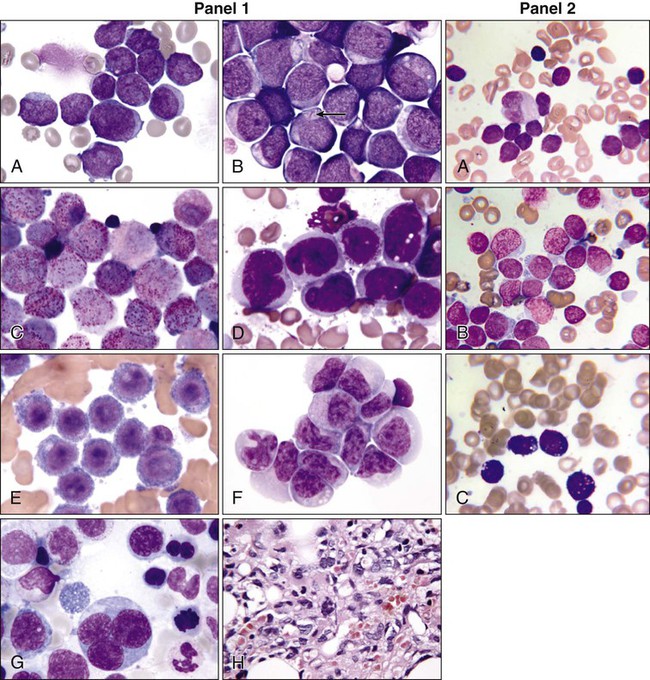
Morphology
Immunophenotyping
AML
Precursor stage
CD34, CD117, HLA-DR
Granulocyte marker
CD13, CD 33, cMPO
Monocytic marker
CD14, CD15, CD11b, NSE
Megakaryocytic marker
CD41a, CD61
Erythroid marker
CD36, CD71
ALL
Pro-B
C19 or CD22+ CD10–
Common ALL
CD10+
Pre-B
Intracytoplasmic Ig+
Mature B
Surface immunoglobulin
Early T
CD7+ CD1a–, surface CD3–
Cortical T
CD1a+, surface CD3–
Mature T
Surface CD3+
Cytogenetics
Risk Status
SWOG15
MRC (1998)16
MRC (2010)17
Favorable
t(15;17), t(8;21), inv(16)/t(16;16)/del(16q)
t(15;17), t(8;21), inv(16)/t(16;16)/del(16q)
t(15;17)(q22;q21), t(8;21)(q22;q22), inv(16)(p13q22)/t(16;16)(p13;q22)
Intermediate
Normal, +8, +6, -Y, del(12p)
Normal, 11q23 abn, +8, del(9q), del(7q), +21, +22, all others
Abnormalities not classified as favorable or unfavorable
Unfavorable
abn(3q), del(5q)/-5, -7/del(7q), t(6;9), t(9;22), 9q, 11q, 20q, 21q, 17p, complex (≥ 3 unrelated abnormalities)
abn(3q), del(5q)/-5, -7, complex (≥ 5 unrelated abnormalities)
abn(3q) [excluding t(3;5)(q21~25;q31~35)], inv(3)(q21q26)/t(3;3)(q21;q26), add(5q)/ del(5q)/-5, add(7q)/del(7q)/-7, t(6;11) (q27;q23), t(10;11)(p11~13;q23), t(11q23) [excluding t(9;11)(p21~22;q23) and t(11;19) (q23;p13)],t(9;22)(q34;q11), -17/ abn(17p),complex (≥ 4 unrelated abnormalities)
Unknown
All other abnormalities
Category not recognized
Category not recognized
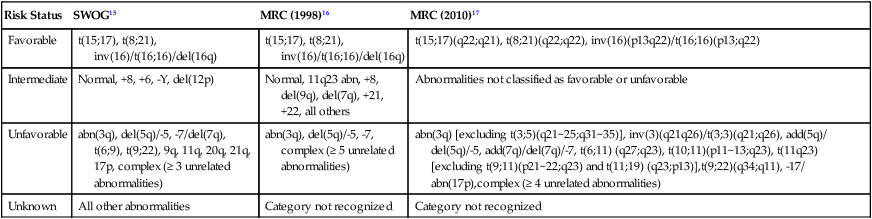
Incidence (%)
FAVORABLE
High hyperdiploidy
10
del 9p
9
UNFAVORABLE
t(9;22)
19
t(4;11)
7
t(8;14)
2
Low hypodiploidy/near triploidy
4
Complex
5
Mutational Events
AML-Associated Mutations
Retinoic Acid Receptor Alpha Translocations
Core Binding Factor Translocations
![]()
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree