History/examination
Infection: fever; localizing symptoms, especially sinus, mouth, anogenital, skin, and lungs
Hemorrhage: petechiae, ecchymosis, epistaxis, oral/GI bleeding, visual complaints
Symptoms of intracranial bleed, including headache and neurologic deficits
Anemia: exertional dyspnea, CHF, angina, orthostasis, syncope
CNS leukemia: headache, confusion, neurologic deficits
Leukostasis: dyspnea, headache, confusion
Monocytic leukemia: gingival hyperplasia and/or nontender cutaneous nodules
Other
Long-standing symptoms suggest preceding myelodysplasia
Full siblings, allergies, major medical problems
HIV risk factors, previous hepatitis
Previous chemotherapy, exposure to benzene
Routine laboratory testing
Before treatment: blood chemistry, LFT, CBC/manual differential, PT/PTT, FDP, fibrinogen, uric acid, LDH, pregnancy test, HIV, HLA class I/II, urinalysis, type/cross
During treatment: CBC daily and blood chemistry/LFT twice weekly
Radiographic studies
Chest radiograph
MUGA scan
Procedures
Placement of central venous access
Lumbar puncture if CNS leukemia is suspected
Leukapheresis if leukostasis is present
Management of a febrile patient
Culture blood, urine, suspected sites of infection
Inspect indwelling line
Initiate broad-spectrum antibiotics promptly
Cefepime or ceftazidine or imipenem
Vancomycin if line infection suspected
Allergic to β-lactams: ciprofloxacin or levofloxacin and vancomycin
Clinically septic: add gentamicin
Oral or possible intra-abdominal infection: add anaerobic coverage
GI, gastrointestinal; CHF, congestive heart failure; CNS, central nervous system; HIV, human immunodeficiency virus; LFT, liver function tests; CBC, complete blood count; PT, prothrombin time; PTT, partial thromboplastin time; FDP, fibrin degradation products; LDH, lactate dehydrogenase; HLA, human leukocyte antigen; MUGA, multiple-gated acquisition.
Bone marrow aspiration and biopsy and cytogenetic studies including conventional karyotype analysis and fluorescent in situ hybridization (FISH), multicolor flow cytometry and cytochemistry studies are essential for diagnosis and classification of acute leukemias. Molecular studies using reverse transcriptase polymerase chain reaction (RT-PCR) for FMS-like tyrosine kinase 3 (FLT3) internal tandem duplication (FLT3-ITD mutation), nucleophosmin gene (NPM-1) mutation, c-Kit mutation, and CCAAT/enhancer binding protein alpha gene (CEBPA) mutation have prognostic importance and therapeutic implications on postremission therapy choices in AML. Although recently supplanted by the World Health Organization (WHO) classification, the French–American–British (FAB) classification remains a commonly used system for the description of acute leukemias. The FAB classification relies on morphology, cytochemistry, and flow cytometry to define subtypes of AML (M0 through M7) and ALL (L1 through L3). Application of these criteria requires a thorough examination of the peripheral blood and bone marrow aspirate with enumeration of blasts, which are characterized by a high nucleus/cytoplasm ratio with fine nuclear chromatin and one or more nucleoli. The FAB criteria define AML when greater than 30% blasts are present in the bone marrow aspirate; greater than 20% blasts define AML in the WHO classification. There are a few exceptions. For diagnosis of acute myelomonocytic leukemia, myeloblasts and promonocytes together should comprise at least 20% of bone marrow cellularity. Acute erythroid leukemia is another exception in which >50% erythroid precursors are present in the bone marrow and myeloblasts should account for >20% of nonerythroid cells. Additionally, t(8;21), inv(16), t(16;16), and t(15;17) are diagnostic of AML regardless of blast percentage in bone marrow. The diagnosis of ALL requires demonstration of at least 20% lymphoblasts in bone marrow. Cytochemistry can be helpful in the diagnosis of acute leukemia (for instance, myeloid blasts are positive for myeloperoxidase), but now flow cytometry is the primary method for determining the leukemia subtype. Lymphoid cells are identified by the presence of CD10, CD19, and CD20 (B cell) or CD2, CD3, CD4, CD5, and CD8 (T cell). Myeloid markers include CD13, CD33, and CD117/c-Kit; CD14 and CD64 (monocytic markers); glycophorin A (erythroid); and CD41 (megakaryocytic).
Subtypes of AML and ALL as defined by the FAB system are of limited prognostic or therapeutic importance. The WHO classification, on the other hand, incorporates cytogenetic and clinical features of known prognostic significance. In the WHO system, AML subtypes are organized into four categories: AML with recurrent genetic abnormalities, AML with multilineage dysplasia (often associated with a preceding myelodysplastic syndrome [MDS]), therapy-related AML, and AML not otherwise categorized (Table 27-2). Therapy-related AML and AML with multilineage dysplasia have a generally poor prognosis, and patients with these subtypes may be candidates for early allogeneic stem cell transplantation. In the WHO classification, ALL is categorized as precursor B-cell ALL or precursor T-cell ALL. Burkitt’s lymphoma/leukemia is grouped with the mature B-cell neoplasms.
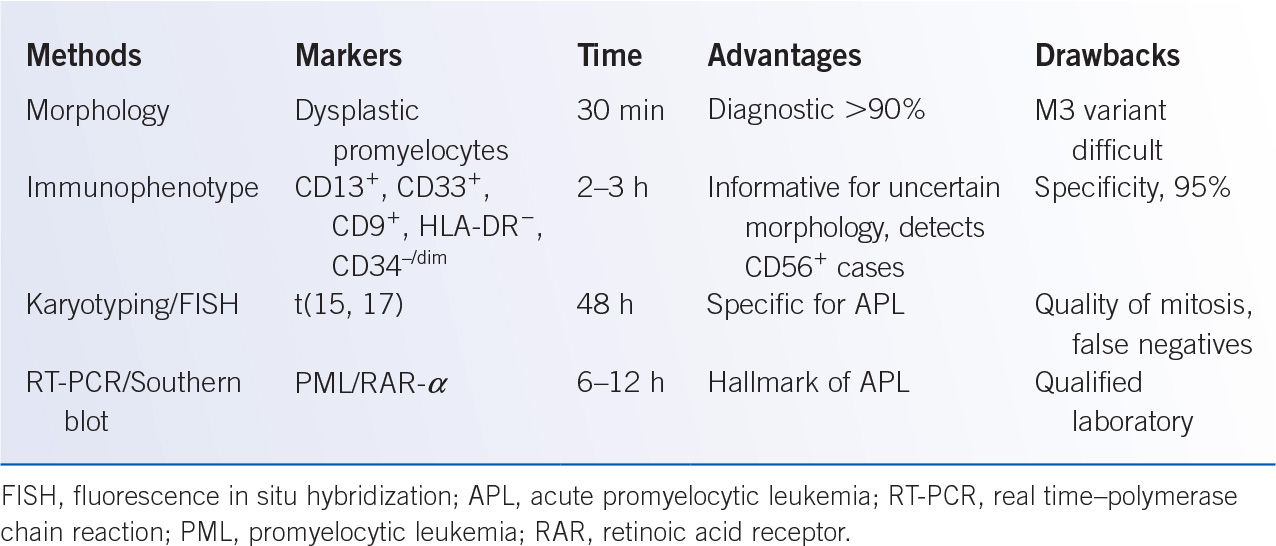
It must be emphasized that cytogenetic studies including conventional karyotype and FISH, and molecular studies for FLT3-ITD, NPM1, c-kit, and CEBPA mutations are crucial for the prognosis and treatment of acute leukemia. Therefore, cytogenetic studies and the above-mentioned molecular studies should be obtained on an initial bone marrow and/or peripheral blood sample. Early recognition of APL (FAB-M3, AML with t[15;17]) is particularly important, as these patients are at risk for DIC, and optimal initial treatment includes all-trans-retinoic acid (ATRA; see Section III.B.). Advantages and pitfalls of diagnostic tools commonly used in the diagnosis of APL are summarized in Table 27-3.
World Health Organization Classification of Acute Myeloid Leukemia |
AML with recurrent genetic abnormalities
t(8;21); (AML1/ETO)
inv(16) or t(16;16); (CBFβ/MYH11)
APL (AML with t(15;17) and variants)
11q23 (MLL) abnormalities
AML with multilineage dysplasia
With a preceding MDS or myeloproliferative disorder
Without a preceding MDS or myeloproliferative disorder
Therapy-related AML and MDS
Related to an alkylating agent
Related to a topoisonerase II inhibitor
AML not otherwise categorized
Minimally differentiated
Without maturation
With maturation
Acute myelomonocytic leukemia
Acute monoblastic and monocytic leukemia
Acute erythroid leukemia
Acute megaloblastic leukemia
Acute basophilic leukemia
Acute panmyelosis with myelofibrosis
Myeloid sarcoma
AML, acute myeloid leukemia; APL, acute promyelocytic leukemia; MDS, myelodysplastic syndrome.
III. THERAPY AND PROGNOSIS. In the absence of antileukemic therapy, the median survival of newly diagnosed patients is 2 to 4 months. Chemotherapy induces responses in most patients with acute leukemia, although for many patients remission is short-lived. The goal of induction chemotherapy is complete remission (CR), which is a prerequisite for cure. CR is defined as normalization of blood counts (ANC more than 1,500/μL, platelet more than 100,000/μL), with bone marrow aspirate/biopsy demonstrating fewer than 5% blasts. Postremission therapeutic options include additional chemotherapy or stem cell transplantation. Chemotherapy given during CR can include consolidation (intensity similar to that for induction) and maintenance (reduced intensity administered for 18 to 36 months).
Advantages and Pitfalls of Diagnostic Tools Commonly Used in the Diagnosis of Acute Promyelocytic Leukemia |
A. AML. As stated above, the prognosis for patients with AML is largely determined by the leukemia risk group. Based on cytogenetic profiles, AML is classified into three risk groups. Molecular abnormalities including FLT3-ITD, NPM1, c-Kit, and CEBPA mutations have significant prognostic impacts. National Comprehensive Cancer Network (NCCN) guidelines have included these mutations in risk classification of AML (Table 27-4). Translocation between chromosomes 8 and 21 (AML-ETO, t[8;21]), present in some patients with FAB-M2 AML, and inversion 16, present in most patients with FAB-M4Eo, have a favorable prognosis with chemotherapy alone. Unfavorable cytogenetics, found in 30% to 40% of AML patients, include monosomies or deletions of chromosomes 5 and 7 (–5, –7, 5q–, 7q–) and equal to or greater than three chromosomal abnormalities. Half of patients with AML have intermediate-risk cytogenetics, primarily a normal karyotype. In a large series from the Cancer and Leukemia Group B (CALGB), the 5-year survival rate was 55%, 24%, and 5% for patients with favorable, intermediate, and unfavorable cytogenetics, respectively.
An active area of investigation is the identification of molecular prognostic factors that can guide therapeutic decisions in patients with normal cytogenetics. One such abnormality is FLT3-ITD mutation, which is present in 20% to 30% of all AML cases and 30% to 40% of normal karyotype AML (NK-AML). FLT3-ITD mutation is associated with a high WBC and a worse prognosis. For instance, among 224 AML patients with normal cytogenetics, 5-year survival was 20% for patients with an FLT3 ITD mutation, versus 42% for those with wild type FLT3. NPM-1 mutations are found in 30% of all AMLs and 50% of NK-AMLs. Isolated NPM-1 mutation in NK-AML is associated with higher CR rates and improved DFS and OS, resulting in outcomes similar to good-risk AMLs. However, patients with both NPM-1 mutations and FLT3-ITD mutations have outcomes more similar to isolated FLT3-ITD mutations. CEBPA mutations can be found in approximately 10% and 15% of all AML patients and patients with NK-AML respectively. NK-AML with CEBPA mutation has a better remission duration and OS similar to core binding factor AML [CBF-AML, another term for AML with t(8:21) and AML with inv(16)]. Double mutation of CEBPA (mutation of both alleles) is less common (5% of NK-AML). A recent study showed that OS benefit of CEBPA mutation is limited to patients with double mutations. This study showed the 8-year OS rates of 54%, 31%, and 34% in double mutation of CEBPA, single mutated CEBPA, and wild type CEBPA respectively.
AML Risk Status by NCCN |
Risk status | Cytogenetics | Molecular abnormalities |
Better-risk | inv(16) or t(16;16) t(8;21) t(15;17) | Normal cytogenetics with NPM1 mutation in the absence of FLT3-ITD or isolated biallelic CEBPA mutation |
Intermediate-risk | Normal cytogenetics +8 alone t(9;11) | t(8;21, inv(16), and t(16;16) with c-KIT mutation |
Poor-risk | Complex cytogenetics (>3 clonal chromosomal abnormalities −5, 5q−, −7, 7q− 11q23—non t(9;11) inv(3), t(3;3) t(6;9) t(9;22) | Normal cytogenetics with FLT3-ITD mutation |
C-Kit mutation is another molecular abnormality with significant prognostic impact in AML with t(8;21) and AML with inv(16). The main mutation clusters are in exon 17 and exon 8. C-kit mutations are seen in approximately 20% to 30% of patients with CBF-AML. In good-risk CBF-AML, c-kit mutation increases the risk of relapse and decreases OS, especially in patients with t(8;21). According to current NCCN guidelines, CBF-AML with c-kit mutation is considered intermediate-risk AML.
For more than 20 years, standard remission induction chemotherapy for AML has included treatment with cytarabine (cytosine arabinoside, ara-C) and an anthracycline. The most common regimen combines 7 days of continuous infusion cytarabine (100 to 200 mg/m2/day) with 3 days of daunorubicin or idarubicin (7 + 3; Table 27-5). A bone marrow examination is repeated 14 to 21 days after starting treatment, and patients with bone marrow cellularity 20% or greater and more than 5% blasts are considered to have residual disease. Patients with persistent disease may achieve a remission after a second, usually abbreviated, course of cytarabine and anthracycline (5 + 2). Sixty percent to 70% of patients achieve a CR after standard induction chemotherapy, with neutrophil (ANC more than 500/μL) and platelet (more than 20,000/μL) recovery occurring an average of 21 to 25 days after the start of therapy. Failure to achieve CR with induction chemotherapy can result from resistant leukemia or early death. Therapy-related mortality increases with age, poor performance status, and underlying organ dysfunction and can be as high as 30% to 40% in elderly patients. Resistant leukemia is associated with a preceding hematologic disorder and adverse cytogenetics, both of which are found more commonly in elderly patients.
Acute Myelogenous Leukemia Chemotherapeutic Regimens |
7 and 3 chemotherapeutic regimen for newly diagnosed AMLa
Ara-C, 100 mg/m2/day, as a continuous infusion for 7 days
Idarubicin 12 mg/m2/day or Daunorubicin, 45–90 mg/m2/day × 3 on days 1, 2, 3 of ara-C
Administration of additional chemotherapy: Perform bone marrow on day 14 of chemotherapy; if cellularity is >20% and blasts are >5%, administer second cycle of chemotherapy (5 and 2): same doses as above with 5 days of ara-C and 2 days of daunorubicin
High-dose ara-C consolidation regimenb
Cytosine arabinoside: 3.0 g/m2 in 500-mL D5W infused i.v. over 3-h period every 12 h twice daily, days 1, 3, 5 (total, six doses)
Before each dose, patients must be evaluated for cerebellar dysfunction; if present, stop drug and do not resume; one way to monitor cerebellar function is to have patients sign name on sheet of paper before each dose; for significant change in signature, physician should evaluate patient before any further therapy is given
To avoid chemical keratitis, administer dexamethasone eye drops, 0.1% 2 drops OU q6h starting 1 h before first dose and continued until 48 h after last dose
APL regimensc
ATRA+ATO regimen:
Remission induction: Daily ATO (0.15 mg/kg) IV, plus oral ATRA (45 mg/m2) until morphologic CR or for a maximum of 60 days
Consolidation therapy: ATO(0.15 mg/kg) IV, 5 days per week, 4 weeks on 4 weeks off, for a total of 4 courses, and ATRA (45 mg/m2) daily 2 weeks on and 2 weeks off for a total of 7 courses
ATRA+Chemotherapy:
Remission induction: IV idarubicin (12 mg/m2/day) on days 2, 4, 6, and 8 plus daily oral ATRA (45 mg/m2) until morphologic CR or for a maximum of 60 days
Consolidation therapy: 3 consolidation courses consisting of idarubicin 5 mg/m2/day on days 1–4 (first cycle), mitoxantrone 10 mg/m2/day on days 1–5 (second cycle), and idarubicin 12 mg/m2 on day 1 (third cycle). Additionally, ATRA 45 mg/m2/day is given simultaneously with chemotherapy from day 1 to day 15 during each consolidation cycle
Maintenance therapy: 2-year maintenance therapy with oral 6-mercaptopurine 50 mg/m2/day, intramuscular methotrexate 15 mg/m2/week alternating with ATRA 45 mg/m2/day given for 15 days every 3 months
AML, acute myelogenous leukemia; ara-C, cytosine arabinoside.
aFrom Bloomfield CD, James GW, Gottlieb A, et al. Treatment of acute myelocytic leukemia: a study by cancer and leukemia group B. Blood 1981;58:1203–1212, and Bob L, Gert JO, Wim van P, et al. High-dose daunorubicin in older patients with acute myeloid leukemia. N Engl J Med 2009;361:1235–1248, with permission.
bFrom Mayer RJ, Davis RB, Schiffer CA, et al. Intensive postremission chemotherapy in adults with acute myeloid leukemia. N Engl J Med 1994;331:896, with permission.
cFrom Lo-Coco F, Avvisati G, Vignetti M, et al. Retinoic acid and arsenic trioxide for acute promyelocytic leukemia. N Engl J Med 2013;369:111–121, with permission.
Patients will invariably relapse if they do not receive additional therapy after achieving CR. In contrast, postremission treatment can result in a cure rate of up to 40%. Commonly used consolidation strategies include high-dose cytarabine (HDAC) or allogeneic stem cell transplantation. HDAC (Table 27-4) can overcome resistance to conventional doses of the drug, producing CR in approximately 40% of patients with resistant leukemia. On this basis, trials of HDAC consolidation for AML in first CR (CR1) were carried out. The value of HDAC consolidation was demonstrated in a CALGB trial, which randomized 596 patients in CR1 to consolidation with four cycles of conventional-dose (100 mg/m2/day × 5 days), intermediate-dose (400 mg/m2/day × 5 days), or high-dose (3.0 g/m2, total of six doses over 5 days) cytarabine. Among patients less than or equal to 60 years old, 4-year progression-free survival was 44% for HDAC consolidation versus 24% for conventional-dose cytarabine. Patients older than 60 did poorly regardless of the type of consolidation they received, with fewer than 20% achieving durable remission. The subgroup analysis of this trial underlines the critical role of cytogenetics for predicting the outcome of consolidation therapy for AML. The estimated likelihood of cure among patients with favorable cytogenetics (t[8;21] and inv16) who received HDAC was 84% versus less than 25% for patients with unfavorable cytogenetics.
Related posts:



Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


