Introduction
Adrenal crisis is the most severe manifestation of adrenal insufficiency (AI); it can be the first manifestation of AI or can occur in patients already established on glucocorticoid therapy. Adrenal crisis is a life-threatening medical emergency associated with high mortality unless it is promptly recognized and treatment is rendered immediately. In this chapter, we discuss the definition, epidemiology, clinical features, etiologies, and management of acute AI. The terms “adrenal crisis” and “acute AI” will be used interchangeably to refer to the same clinical entity.
DEFINITION AND PATHOPHYSIOLOGY
Adrenal crisis occurs when circulating adrenal steroid hormones are insufficient for physiologic requirements. This can arise due to an acute decrease in cortisol synthesis as a result of dysfunction or destruction of the adrenal glands themselves, or due to impaired pituitary secretion of adrenocorticotropic hormone (ACTH) that is responsible for signaling to the adrenal glands to produce cortisol. It can also occur if the body is unable to appropriately increase endogenous cortisol production during times of increased physiologic demand, such as during major surgery, injury, trauma, or infection. Adrenal crisis thus results in an acute deterioration in clinical status associated with absolute or relative hypotension; the hypotension and associated symptoms improve rapidly (within 1 to 2 hours) on parenteral glucocorticoid administration.
EPIDEMIOLOGY
Adrenal crises have an estimated incidence of 5 to 10 cases per 100 patient years and are responsible for increased morbidity and mortality in patients with AI. Each year, about 6% to 8% of patients with AI are reported to suffer an episode of adrenal crisis. About 40% of chronic AI patients have been reported to experience at least one adrenal crisis in their lifetime, and about 20% of chronic AI patients have experienced more than one adrenal crisis in their lifetime. Adrenal crisis is slightly more common in patients with primary AI than in patients with secondary AI. Women make up about 60% of patients admitted with adrenal crisis, which reflects the increased prevalence of AI in women due to an increased predisposition to autoimmune disease. , A prospective study reported 0.5 deaths per 100 patient years from adrenal crisis.
The most common causes of AI are outlined in Table 14.1 . , , Primary AI is most commonly due to autoimmune adrenalitis (Addison’s disease) in the United States and other first-world countries. This can occur as isolated Addison’s disease (in 40% of cases) or as part of an autoimmune polyglandular syndrome (in 60% of cases). In other parts of the world, infections (tuberculosis, HIV, cytomegalovirus [CMV], systemic fungal infections) remain the most common cause. Less frequent causes of primary AI include adrenal metastases, infiltrative diseases, hemorrhage, congenital adrenal hyperplasia, bilateral adrenalectomy, and drugs that impair glucocorticoid production and action. Of note, metastases and infiltrative diseases rarely cause AI because extensive damage to both adrenal glands is required to cause AI.
| Type | Common Causes |
|---|---|
| Primary Causes of AI | |
| Autoimmune |
|
| Infections |
|
| Metastasis |
|
| Adrenal hemorrhage |
|
| Infiltration |
|
| Bilateral adrenalectomy |
|
| Congenital adrenal hyperplasia |
|
| Drug induced |
|
| Other |
|
| Secondary Causes of AI | |
| Pituitary pathologies and insults |
|
| Suppression of HPA axis |
|
| HPA, Hypothalamic-pituitary-adrenal. | |
Secondary AI is more common than primary AI and is caused by any process that leads to deficiency of ACTH. This is most frequently due to pituitary pathologies such as mass lesions, surgery, trauma, medications, and radiation-induced pituitary damage. Secondary AI can also be caused by the chronic use of glucocorticoids, opioids, or other drugs (e.g., checkpoint inhibitors). Exogenous glucocorticoid use by any route (oral, inhaled, topical, or injected) at supraphysiologic doses (prednisone 5 mg or higher per day, or its equivalent) for longer than 4 weeks is well known to suppress the hypothalamic-pituitary-adrenal axis.
Clinical Presentation
RECOGNIZING ACUTE ADRENAL INSUFFICIENCY
The cardinal manifestation of acute AI is absolute or relative hypotension, which ultimately leads to shock if untreated. , , In addition to the defining feature of hypotension, patients with adrenal crisis frequently present with fever and acute abdominal pain. They may also exhibit milder symptoms of hypocortisolism such as fatigue, nausea, anorexia, myalgias, arthralgias, and postural dizziness. The clinical features of an adrenal crisis are compared and contrasted with those of chronic AI in Table 14.2 . , Without hypotension or evidence of hemodynamic compromise, the presence of these other symptoms in a patient with a known diagnosis of AI should be considered distinct from, but a likely precursor to, an adrenal crisis. Failure of hypotension to respond to vasopressor agents should be a clue to the possible presence of acute AI and an indication for the trial of glucocorticoid therapy. Prompt resolution of these features after parenteral glucocorticoid administration is a key feature of adrenal crisis. If the symptoms and hypotension attributed to an adrenal crisis fail to improve within 1 to 2 hours of sufficient glucocorticoid administration, alternative or coexisting diagnoses such as sepsis should be considered.
| Chronic AI | Acute AI (Adrenal Crisis) | |
|---|---|---|
| Symptoms | Fatigue, anorexia, weight loss, myalgias, arthralgias Dizziness Nausea, vomiting, diarrhea Salt craving (in primary AI only) | Severe weakness Acute abdominal pain Nausea, vomiting Syncope Confusion |
| Signs | Orthostatic hypotension Fever Hyperpigmentation of skin creases and buccal mucosa (in primary AI only) | Hypotension Fever Abdominal tenderness, guarding Reduced consciousness, delirium |
| Laboratory Findings | Hyponatremia Hyperkalemia (in primary AI only) Hypoglycemia Hypercalcemia | Hyponatremia Hyperkalemia (in primary AI only) Hypoglycemia Hypercalcemia |
Due to its presentation with hypotension and fever, an adrenal crisis can initially lead clinicians to suspect infection. Patients with fever should indeed be treated as though they have an infection until proven otherwise. Infections are commonly the precipitating events of adrenal crisis. , Bacterial infections are common precipitants of adrenal crisis in elderly patients. Gastroenteritis is a frequently cited trigger that can cause a particularly severe presentation due to rapid dehydration and an inability to tolerate oral medications and fluids. , , Furthermore, the combination of abdominal pain, tenderness, and fever may mimic “acute abdomen,” and abdominal exploration in such patients can be catastrophic.
Several biochemical abnormalities can be seen in patients with acute AI, although none distinguish adrenal crisis from chronic AI, as summarized in Table 14.2 . Hyponatremia is the most common biochemical abnormality, occurring in 70% to 80% of cases. Hyponatremia can be seen in patients with any etiology of AI due to increased vasopressin secretion resulting from cortisol deficiency, although it is more common in primary AI in which mineralocorticoid deficiency compounds the problem via natriuresis and volume depletion. , Patients with primary AI can have hyperkalemia due to mineralocorticoid deficiency. Hypoglycemia may be seen in AI of any etiology due to glucocorticoid deficiency causing impaired gluconeogenesis. Hypoglycemia is more commonly seen in children with adrenal crisis than in adults. It is also more common in cases of secondary AI caused by isolated ACTH deficiency. , In a minority of cases of adrenal crisis, patients may have the additional laboratory abnormalities of hypercalcemia (thought to occur due to hypovolemia, acute kidney injury, and decreased renal excretion of calcium) and normocytic anemia (cortisol is required for the maturation of blood progenitor cells and pernicious anemia may coexist in autoimmune polyglandular syndromes).
PRIMARY ADRENAL INSUFFICIENCY
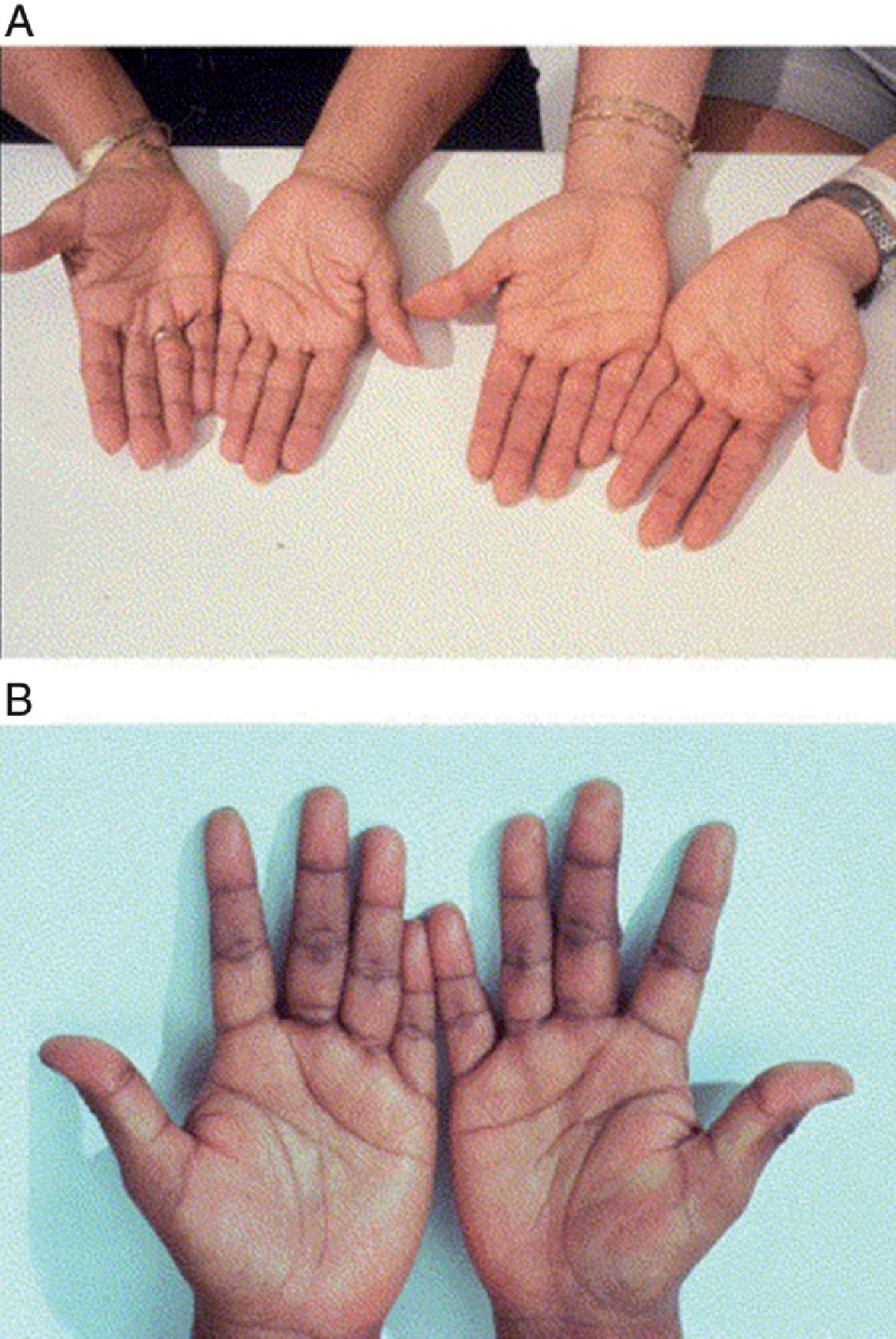
Certain features of a patient’s presentation with acute AI vary according to etiology and can serve as clues to the etiology of AI ( Table 14.3 ). As mentioned previously, hyponatremia and especially hyperkalemia indicate primary AI. Patients with chronic primary AI who present in crisis may have hyperpigmentation, which occurs due to the chronic hypersecretion of ACTH that is cosecreted with melanocyte-stimulating hormone (MSH). This hyperpigmentation is seen in 41% to 74% of cases, and is most noticeable in palmar creases and buccal mucosa, as demonstrated in Figs. 14.1 and 14.2 . , , ,
| Clinical Feature | Potential Etiology | |
|---|---|---|
| History | Personal or family history of autoimmune disease | Addison’s disease (autoimmune adrenalitis) – classical primary AI |
| Salt craving | Primary AI | |
| Recent induction of general anesthesia or rapid-sequence intubation | Primary AI due to etomidate | |
| On anticoagulant therapy | Bilateral adrenal hemorrhage | |
| Coagulopathy | Bilateral adrenal hemorrhage | |
| Abdominal or back trauma | Bilateral adrenal hemorrhage | |
| Post-partum hemorrhage | Sheehan syndrome | |
| Acute headache | Pituitary apoplexy | |
| On immune checkpoint inhibitor therapy, especially CTLA-4 inhibitor or combination therapy | Hypophysitis | |
| Signs | Hyperpigmentation | Chronic primary AI |
| Vitiligo | Addison’s disease (autoimmune adrenalitis) – classical primary AI | |
| Petechial rash | Waterhouse-Friderichsen syndrome | |
| Nuchal rigidity, delirium | Meningococcal meningitis | |
| Abdominal tenderness, guarding | Bilateral adrenal hemorrhage | |
| Peripheral visual field deficit | Pituitary apoplexy | |
| Laboratory Findings | Hyperkalemia | Primary AI |
| Hypoglycemia | Secondary AI (more common) > Primary AI |

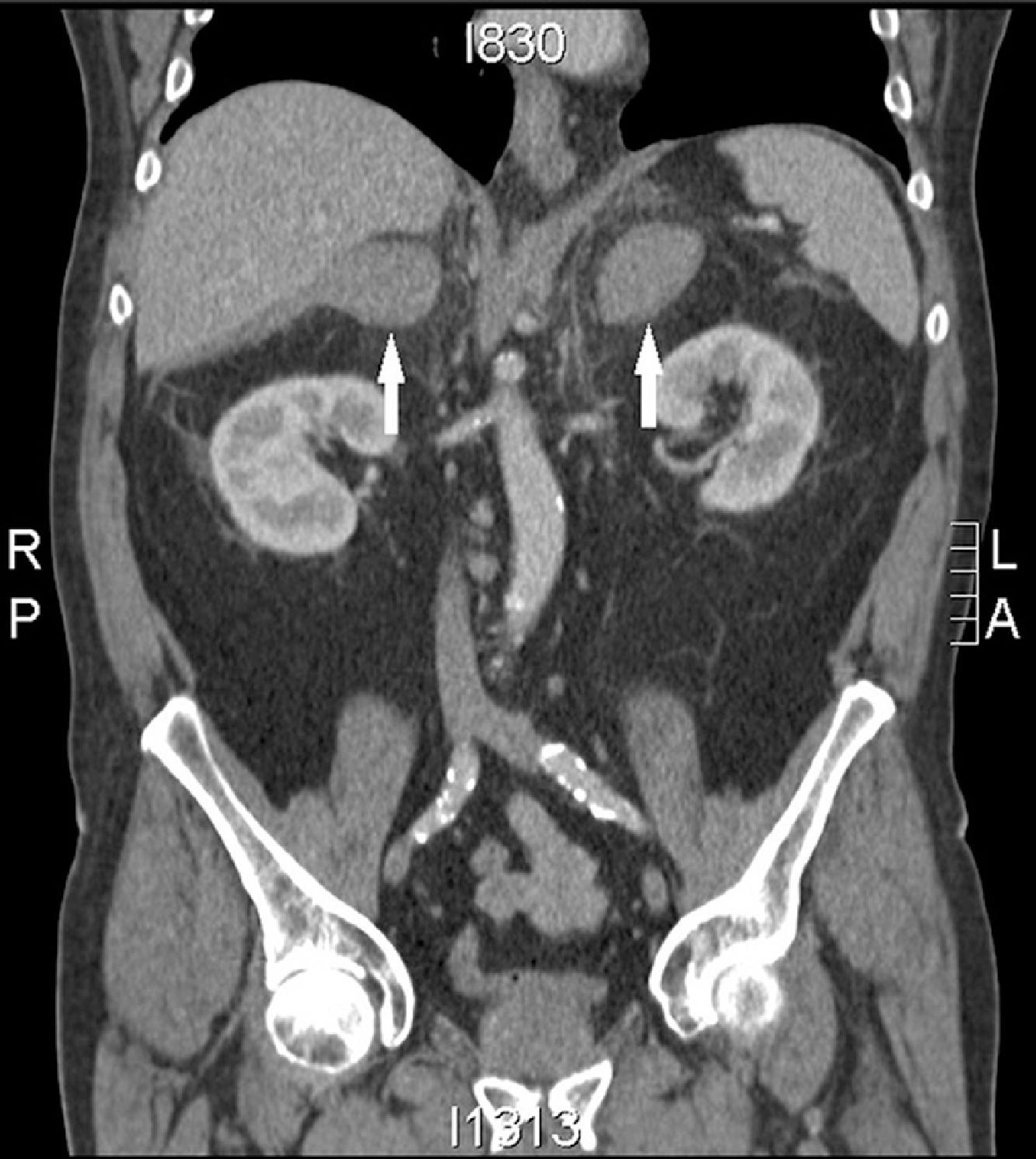
An important etiology of acute AI is bilateral adrenal hemorrhage and infarction. In contrast to patients with chronic primary AI, these patients do not have hyperpigmentation due to acuity of the changes (lack of prolonged elevation of ACTH and cosecreted MSH). The majority of patients with bilateral adrenal hemorrhage have abdominal, flank, back, or lower chest pain. These patients can rapidly deteriorate to shock without preceding hypotension. Risk factors for adrenal hemorrhage include thromboembolic disease, anticoagulant therapy, underlying coagulopathy (such as antiphospholipid antibody syndrome or disseminated intravascular coagulation), blunt trauma, and postoperative state. When a patient with such risk factors develops changes such as fever, hypotension, or abdominal pain, clinicians should promptly suspect adrenal crisis due to hemorrhage. A noncontrast computed tomography (CT) scan can confirm the diagnosis, with acute adrenal hemorrhage appearing as bilaterally enlarged, hyperdense adrenal glands, as shown in Fig. 14.3 .
Various infections have also been associated with acute hemorrhagic necrosis of the adrenal glands, which is known as Waterhouse–Friderichsen syndrome (WFS) and is more common in children than in adults. This may be typically caused by bacterial infection, and has classically been described in the context of meningococcemia in the prevaccine era. However, WFS has also been reported due to sepsis from other bacterial pathogens such as Haemophilus influenzae , Pseudomonas aeruginosa , Escherichia coli, Mycoplasma pneumoniae, Streptococcus pneumoniae, and Staphylococcus aureus . Viral infections including CMV, parvovirus B19, varicella zoster, and Epstein–Barr virus have also been reported to cause WFS. Physical findings in patients with WFS include petechial rash, purpura fulminans, and neurologic changes in meningitis, in addition to typical signs of infection.
Finally, several drugs can precipitate adrenal crisis. Mechanisms of drug-induced primary AI include an inhibition of enzymes involved in cortisol biosynthesis (e.g., ketoconazole, metyrapone, etomidate) and an increase in glucocorticoid metabolism (e.g., carbamazepine, rifampin, phenytoin). Additionally, initiation of thyroxine therapy (thyroid hormone) in hypothyroid patients with undiagnosed AI may potentially precipitate acute AI, as thyroid hormone may increase the clearance of cortisol. , , This is particularly important to consider because patients with AI often have concomitant hypothyroidism, either as part of autoimmune polyglandular syndrome type 2 (primary autoimmune AI and hypothyroidism) or due to pituitary disease (secondary/central AI and hypothyroidism).
SECONDARY ADRENAL INSUFFICIENCY
Acute AI is slightly less frequent in patients with secondary AI, likely because these patients have partial preservation of cortisol secretion, and because mineralocorticoid secretion is preserved. Still, adrenal crisis can occur when there is acute loss of pituitary function causing acute hypocortisolism, such as in cases of pituitary apoplexy (sudden hemorrhage, usually occurring into a pituitary macroadenoma), infarction of the pituitary gland after post-partum hemorrhage (Sheehan syndrome), during pituitary surgery, or head trauma. The majority of patients with pituitary apoplexy have headache and visual field deficit. Other pituitary axes may be compromised as well.
Immune checkpoint inhibitor therapy, which is increasingly used in the treatment of melanoma and other cancers, can cause acute AI, either due to hypophysitis causing secondary AI or due to adrenalitis causing primary AI. In a large metaanalysis, the incidence of hypophysitis in patients on checkpoint inhibitor therapy was found to be 6.4% with combination therapy, 3.4% with CTLA-4 inhibitors, 0.4% with PD-1 inhibitors, and less than 0.1% with PD-L1 inhibitors. A separate study found that in patients with hypophysitis due to checkpoint inhibitor therapy, secondary AI was the most common anterior pituitary deficiency, present in 83% of cases. Primary AI is less frequently observed, with 4.2% incidence in patients receiving combination therapy and 0.7% with any checkpoint inhibitor.
Patients with chronic exogenous glucocorticoid use can present with acute secondary AI when the glucocorticoid therapy is abruptly discontinued, either due to patient nonadherence or clinician error. Such patients may have physical signs of Cushing’s syndrome such as wide violaceous striae, dorsocervical and supraclavicular fat pads, proximal muscle wasting, thin skin, ecchymoses, and central adiposity. However, one study in Australia found that adrenal crises were rare in patients with glucocorticoid-induced AI.
Chronic opioid use is an increasingly important cause of secondary AI to recognize in the modern era of the opioid epidemic. Among patients treated with chronic opioids, the estimated prevalence of opioid-induced AI ranges from 9% to 29%. Clinically significant adrenal crisis in patients with opioid-induced AI has been described in some case reports. ,
Diagnosis
In the hemodynamically unstable patient in whom adrenal crisis is clinically suspected, therapy should be initiated promptly prior to biochemical confirmation of the diagnosis. Thus, having a high index of suspicion for this condition based on patient history and clinical presentation is of the utmost importance. When acute AI is suspected, blood should be drawn for serum cortisol and electrolyte testing, ideally just prior to giving the first dose of glucocorticoid. Some experts also recommend drawing and holding blood samples for ACTH, renin, and aldosterone, to be processed later if the diagnosis of AI is likely.
A serum cortisol of less than 5 μg/dL during a period of acute stress or illness (regardless of time of day) strongly supports the diagnosis of AI. Conversely, a serum cortisol concentration greater than 20 μg/dL excludes the diagnosis. In cases in which the serum cortisol is indeterminate (5 to 20 μg/dL), glucocorticoid therapy should be continued until the patient has recovered, and further testing (which may include an ACTH stimulation test) can be conducted later to clarify the diagnosis. There is no role for ACTH stimulation testing during an acute hemodynamic decompensation that is suspected to be due to AI.
If the cortisol concentration is low, suggesting the diagnosis of AI, then the clinician should use additional laboratory tests to determine the level of the defect. Elevated ACTH concentration indicates primary AI, and in these cases, one would expect low aldosterone with elevated renin. Low or inappropriately normal ACTH concentrations indicate secondary AI; aldosterone and renin would be normal in these cases. Further testing, such as imaging of the adrenal glands (using CT) or pituitary (using magnetic resonance imaging), may be obtained as clinically indicated to determine the specific etiology of the AI.
A caveat to interpretation of cortisol concentrations in critically ill patients is that patients may have abnormalities of binding proteins (cortisol-binding globulin, albumin) that affect the total serum cortisol (which is routinely measured) but do not affect the free cortisol (which is biologically active). Testing the free cortisol concentration in saliva or serum has been suggested but is not routinely performed or recommended due to the lack of availability, expense, and lack of validity and established criteria for interpretation. In lieu of testing the free cortisol concentration, clinicians should measure total cortisol and albumin, and consider conditions that may alter binding protein levels in a patient, such as liver cirrhosis, nephrotic syndrome, and estrogen use (e.g., in oral contraceptives). Also, physiologic states like pregnancy may result in elevated total cortisol levels due to increased cortisol-binding globulin.
In addition to suspecting and diagnosing acute AI, clinicians should perform a thorough evaluation for the precipitating cause, which usually includes reviewing the patient’s medications, testing for infection, and obtaining imaging as indicated.
Management of Acute Adrenal Insufficiency
Treatment of patients with possible adrenal crisis should not be delayed while diagnostic tests are being completed. Prompt treatment includes immediate parenteral administration of hydrocortisone 100 mg; if intravenous access is not available, intramuscular injection can be given. Concurrently, it is necessary to administer aggressive fluid resuscitation of 1 L of isotonic saline with or without 5% or 10% glucose (depending on the presence of hypoglycemia) in the first hour. Then, hydrocortisone is given intravenously either via continuous infusion to equal 200 mg in a 24-hour period or 50 mg intravenously every 6 hours in 24 hours. Intravenous isotonic saline is continued based on the patient’s hemodynamic stability, volume status, and urine output. ,
If hydrocortisone is not available, prednisolone could be used. Dexamethasone is the least favorable steroid to use, especially in adrenal crisis due to primary AI, because it has no mineralocorticoid activity, whereas hydrocortisone 40 mg has an equivalent of 100 μg of fludrocortisone.
Blood gas and glucose should be monitored hourly until acidosis and hypoglycemia resolve and then every 2 to 4 hours thereafter. Electrolytes should be monitored every 4 hours. Caution is recommended to not exceed correction of hyponatremia greater than 10 to 12 mEq/L in the first 24-hour period due to the risk of central pontine myelinolysis. Cortisol replacement can induce water diuresis, and in cases of secondary AI can also suppress antidiuretic hormone. In combination with isotonic fluid replacement, these changes can lead to overcorrection of hyponatremia and osmotic demyelination syndrome. Hyperkalemia usually normalizes with fluid, electrolyte, and steroid replacement. Cardiac monitoring should be in place to assess for any ECG changes (peaked T waves, widened QRS complexes, and/or flattened P waves) due to hyperkalemia.
The identification and treatment of the illness or injury that precipitated the adrenal crisis is also important.
On day 2, if clinically improved, hydrocortisone could be decreased to 100 mg/24-hour period (i.e., 25 mg every 6 hours). As the patient continues to improve, the patient can be transitioned from parenteral to oral glucocorticoids.
Chronic Management of Adrenal Insufficiency and Prevention of Adrenal Crisis
The optimal glucocorticoid maintenance regimen is hydrocortisone 15 to 25 mg/day or cortisone acetate 20 to 35 mg/day. This should be divided into twice daily or three times daily doses with the highest dose in the morning and the lowest in the afternoon (not later than 4 to 6 hours before bedtime) in order to mimic endogenous glucocorticoid pulsatile circadian release, which reaches its highest peak in the morning. Hydrocortisone and prednisolone are active glucocorticoids, whereas prednisone and cortisone acetate are prodrugs that require activation via 11β-hydroxysteroid dehydrogenase type 1 in the liver before exerting their activity.
Patients with primary AI need mineralocorticoid replacement to maintain water, electrolyte, and blood pressure homeostasis. Mineralocorticoid replacement is given in the form of fludrocortisone at initial doses of 50 to 100 µg in adults. Patients are recommended to not restrict the salt in their diet.
Patient education, such as increasing dose of replacement therapy (“stress dosing”) when sick or planned for a medical procedure/surgery, using parental hydrocortisone as necessary, and seeking medical assistance promptly, is the best approach to prevention of adrenal crisis. , , Follow-up care with an endocrinologist is necessary to periodically reevaluate for symptoms of inadequate glucocorticoid replacement (symptoms of AI) or for signs of excessive glucocortocoid replacement (features of Cushing’s syndrome). The endocrinologist should monitor for adverse metabolic effects of excessive glucocorticoid therapy, such as weight gain, hyperglycemia, dyslipidemia, and low bone density. The adequacy of mineralocorticoid replacement should be assessed by measuring orthostatic vital signs, serum potassium, and plasma renin activity. Additionally, the endocrinologist should counsel patients to wear a medical alert identification and reeducate patients at each visit about how to recognize adrenal crisis, when and how to increase glucocorticoid dosing during illness, how to self-administer parenteral glucocorticoid, and when to seek emergent medical care. ,
References
Related posts:



Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


