Fig. 16.1
ALDHs modulate carcinogenesis by metabolizing acetaldehyde and retinaldehyde. Ethanol is metabolized by alcohol dehydrogenase (ADH), catalase, and CYP2E1 to acetaldehyde. Acetaldehyde can interfere with antioxidative defense systems (AODS) and generate reactive oxygen species (ROS); inhibits DNA repair and methylation; and forms DNA and protein adducts to promote tumor growth. Acetaldehyde is metabolized to acetate primarily by ALDH2, ALDH1B1, and ALDH1A1. Retinaldehyde, formed from retinol by ADH, is converted to retinoic acid (RA) by retinaldehyde-metabolizing ALDHs. RA exerts anticarcinogenic activity by binding to cellular retinoic acid-binding proteins (CRBPII) and activating the RA receptor (RAR). When RA binds to fatty acid-binding protein 5 (FABP5), it activates orphan nuclear receptor peroxisome proliferator-activated receptor (PPAR)β/δ and acts as procarcinogenic agent. ALDH aldehyde dehydrogenase, NAD + NAD(P), nicotinamide adenine dinucleotide (phosphate), H 2 O 2 hydrogen peroxide
Table 16.1
ALDH expression in various progenitor, stem, and cancer cell types
S.No. | Cell or tumor type | ALDH isozyme(s)a | Reference |
|---|---|---|---|
1 | Hematopoietic progenitor | ALDH, ALDH1A3 | |
2 | Mesenchymal progenitors | ALDH | [75] |
3 | Endothelial progenitors | ALDH | [75] |
4 | Neural stem cells | ALDH, ALDH1L1 | |
5 | Normal mammary stem cells | ALDH1A1 | [26] |
6 | Breast cancer stem cells | ALDH1A1, ALDH1A3, ALDH2, ALDH6A1 | |
7 | Prostate cancer | ALDH, ALDH7A1 | |
8 | Ovarian cancer stem cells | ALDH, ALDH1A1 | |
9 | Ovarian cancer cells | ALDH1A1, ALDH1A3, ALDH3A2, ALDH7A1 | [83] |
10 | Colon stem cells | ALDH1A1, ALDH1B1 | |
11 | Colon cancer stem cells | ALDH1A1, ALDH1B1 | |
12 | Leukemia stem cells | ALDH | [20] |
13 | Human lung cancer cells | ALDH1A1 | |
14 | Head and neck cancer stem cells | ALDH1A1 | [87] |
15 | Pancreatic cancer | ALDH, ALDH1A1, ALDH1A3 | |
16 | Liver cancer stem cells | ALDH, ALDH1A1 |
A causal relationship exists between alcohol consumption and CRC or pancreatic cancer and this may be mediated, at least in part, by acetaldehyde [12, 15]. The significance of retinaldehyde and acetaldehyde in tumor formation, and very high expression of the ALDHs in colorectal and pancreatic cancer are suggestive of a crucial role for acetaldehyde- and retinaldehyde-metabolizing ALDHs in these cancers. Lack of ALDH2 activity and resultant high acetaldehyde levels are linked with colon cancer initiation. By contrast high ALDH1 activity (primarily ALDH1A1 and ALDH1B1) is required for the stemness and tumorigenic potential of CSCs.
16.2 Acetaldehyde: A carcinogen
Acetaldehyde is categorized as “carcinogenic to humans” and “reasonably anticipated to be a human carcinogen” according to IARC regulations and US National Toxicology Program (NTP), respectively [13, 33]. Acetaldehyde has been shown to be a highly toxic, mutagenic, and carcinogenic compound in a variety of in vitro and in vivo studies. Its effects range from damaging antioxidant defenses [11] to interfering with DNA methylation and repair mechanisms through formation of adducts with DNA and proteins (Fig. 16.1) [10, 12]. In the colon, acetaldehyde is primarily produced from ethanol by resident bacteria and, to a lesser extent, by mucosal alcohol dehydrogenase (ADH). As a result of metabolism by intra-colonic microbes, large quantities (nine-fold higher than normal) of acetaldehyde accumulate in the rat colon 2 h after intraperitoneal injection of ethanol [34]. Human colon mucosal cells harbor ADH1, ADH3, and ADH5, with the ADH1 and ADH3 isozymes being most active [35]. In an in vitro experiment, human colon contents were able to generate 60–250 μM acetaldehyde when incubated with concentration of ethanol (10–100 mg%), which is known to be attained during normal ethanol drinking [36]. The high levels of acetaldehyde attained in the colon after drinking ethanol likely underlies the correlation between chronic, heavy ethanol consumption, and CRC in humans. In ethanol-treated rats, a high concentration of acetaldehyde (50–350 μM) in the colon mucosa has been shown to correlate positively with hyper-proliferation of the colon crypt cells. Such a phenomenon would be anticipated to favor the development of CRC [37, 38].
Acetaldehyde is metabolized primarily by mitochondrial ALDH2 and ALDH1B1 and, to lesser extent, by cytosolic ALDH1A1 (Table 16.2) [14]. The most convincing evidence for a role of acetaldehyde in CRC initiation emanates from studies involving Asians who possess a polymorphism in their ALDH2 enzyme known as ALDH2*2. These subjects possess a single nucleotide polymorphism (SNP) that leads to a lysine to glutamate substitution at residue 487 that renders the enzyme functionally inactive [39, 40]. Approximately 40 % of the Asian population carry an ALDH2*2 allele; this compromises their ability to metabolize acetaldehyde and increases their colon cancer risk 3.4 times [16].
Table 16.2
Affinity of ALDHs for acetaldehyde and retinaldehyde
S.No. | ALDH isozyme(s) | Substrate | K m (μM) | Reference |
|---|---|---|---|---|
1 | ALDH1A1 | Acetaldehyde | 180 | [14] |
All-trans retinaldehyde | 11.6–26.8 | [91] | ||
9-cis retinaldehyde | 3.59 | Jackson et al., under preparation | ||
2 | ALDH1A2 | All-trans retinaldehyde | 0.66 | [92] |
9-cis retinaldehyde | 0.62 | |||
3 | ALDH1A3 | All-trans retinaldehyde | 0.2 | [93] |
4 | ALDH1B1 | Acetaldehyde | 55 | [14] |
Retinaldehyde | 24.9 | Jackson et al., under preparation | ||
5 | ALDH2 | Acetaldehyde | 3.2 | [14] |
6 | ALDH8A1 | 9-cis retinaldehyde | 3.15 | [46] |
16.3 Opposing Effects of Retinoic Acid on Cancer Cell Proliferation
Retinoids exert many physiologically important and diverse functions in relation to cellular proliferation and differentiation of normal and cancer cells. For example, retinoids are crucial for embryonic development and adult tissue remodeling. The retinoids comprise all of the derivatives of retinol, including all-trans-, 9-cis-, and 13-cis– retinoic acid (RA). Retinol is oxidized to retinaldehyde by retinol dehydrogenases. The resultant retinaldehydes are further metabolized to their corresponding RA by retinaldehyde dehydrogenases which include RALDH1 (ALDH1A1), RALDH2 (ALDH1A2), RALDH3 (ALDH1A3), and RALDH4 (ALDH8A1) (Table 16.2) [41–46]. Among the RAs, all-trans-RA (ATRA) is the most biologically potent retinoid. Abnormally low levels of ALDH1A2 have been observed in breast and prostate cancers [47, 48]. Impaired RA formation and high levels of CYP26A1 (an RA-metabolizing enzyme) in human breast cancer are consistent with a protective role for RA in this cancer [47–49]. The physiological actions of the retinoids are mediated through binding of the RA receptor (RAR) and retinoid X receptor (RXR) heterodimer to the regulatory region of retinoid-responsive genes, known as RA response elements [50]. RARs and RXRs are ligand-dependent transcription factors and exist as α, β, or γ isoforms. RAR isoforms interact with both ATRA and 9-cis RA, whereas RXR isoforms interacts only with 9-cis RA [51, 52]. The binding of RA with the RAR/RXR dimer recruits co-activator proteins and initiates transcriptional activation of the retinoid-responsive genes [50]. Retinoids have been found to be effective for the treatment of acute promyelocytic leukemia and prevention of liver, lung, breast, prostate, skin, and colon cancers [53–55]. In vivo studies involving rats have revealed that retinoids added to the diet reduced colon cancer cell proliferation and prevented azoxymethane-induced aberrant crypt foci (putative precancerous lesions in colon) and colon tumor formation [55, 56]. An RXR-selective retinoid, AGN194204, has been found to inhibit the proliferation of human pancreatic cancer cells, an effect that can be reversed by an RXR-selective antagonist [57]. In addition to inhibiting the growth of pancreatic cancer cells, RA increases the sensitivity of pancreatic adenocarcinoma cells to the antineoplastic drugs gemcitabine and cisplatin [58].
In contrast to the antiproliferative and anti-survival role of RA in cancer cells, dietary ATRA has been shown to enhance initiation and growth of intestinal tumors in the Apc(Min)/+ mouse model in vivo [59]. RA can promote cell survival and hyperplasia in cells expressing high levels of fatty acid-binding protein 5 (FABP5) by activating an orphan nuclear receptor, peroxisome proliferator-activated receptor (PPAR)β/δ [60]. PPARβ/δ mediates antiapoptotic properties partly by inducing the PDK1/Akt survival pathway [61]. RA binds to intracellular lipid-binding proteins (iLBPs), including cellular retinoic acid-binding proteins (CARBPII and FABP5). CARBPII and FABP5 are selective for nuclear receptors RARα and PPARβ/δ, respectively [60]. Hence, RA induces CARBPII- or FABP5- mediated activation of RAR or PPARβ/δ (respectively), depending on the ratio of FABP5/CRBPII in the cells [60]. Human CRC cell lines (specifically, T84, COLO205, SW620, SW480, HCT116, and DLD-1) express ~30-fold higher levels of FABP5 relative to normal colorectal cells (CCD18-Co), suggesting the possibility of pro-proliferative and antiapoptotic roles for RA in these cells [60, 62]. However, the expression levels of PPARβ/δ in CRC cells and its role in tumorigenesis are unresolved in various cancers, including CRC [63].
RA inhibits the proliferation and increases chemosensitivity of pancreatic cancer cells. However, the involvement of RA in CRC is less clear, with opposing findings suggesting pro- or antiproliferative roles.
16.4 ALDHs and Cancer Stem Cells
In the gastrointestinal (GI) tract, tissue-specific stem cells are at the top of the cellular hierarchy and play a critical role in regulating tissue homeostasis. These specialized epithelial cells are characterized by their ability to self-renew and differentiate into a variety of cellular populations that perform specific functions within the GI tract. Currently, it is believed that these tissue-specific stem cells (or progenitor cells), when oncogenically transformed, become CSCs or tumor-initiating cells (TICs) since they functionally possess the capacity to form tumors and maintain tumor growth. Accumulating evidence also suggests that CSCs are responsible for chemotherapeutic/radiation resistance and tumor recurrence (Fig. 16.2). ALDH catalytic activity has been identified in many human cancers [28] and, as such, is used as a marker of CSCs, including colorectal and pancreatic cancer. The pathophysiological function of ALDHs in CSCs remains unresolved. Intense research of ALDH enzymes is underway in order to elucidate the role of these proteins in the development and progression of cancer as well as drug resistance.
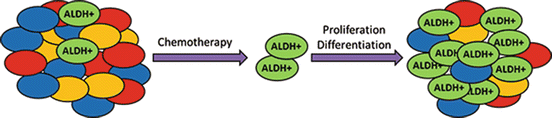
Fig. 16.2
ALDH-expressing cells are responsible for chemoresistance and relapse of many tumors after chemotherapy. Most current chemotherapy drugs are effective against the bulk of the tumor cells. However, the high ALDH-expressing (ALDH+) cancer stem cells are resistant to these treatments. As a result, during chemotherapy, the ALDH+ cells proliferate and promote tumor growth. The resultant tumors contain an increased proportion of ALDH+ cells making them more resistant to chemotherapy than the original tumor
16.4.1 Colorectal Cancer
Although earlier stages of CRC are highly curable, therapeutic interventions in advanced disease have proven to be poorly effective at increasing the 5-year survival rate. Recent drug development has focused on targeting the CSC population as a potential therapy. In normal colon, CSCs reside at the bottom of the crypt and generate upward, migrating and differentiating transit amplifying cells (in the middle of the crypt) which become terminally differentiated cells as they move upward and eventually shed into the lumen (Fig. 16.3a) [64]. In CRC, several different molecules, including the cell surface markers CD133 and CD44 as well as ALDH activity, have been proposed as biomarkers for identification and isolation of the CSC population [31, 65–68]. CD133+ colon cancer cells were initially shown to be tumorigenic [67, 68]. However, subsequent studies identified that both CD133+ and CD133− cells possess tumorigenic potential [69]. CD44+ (either with or without epithelial-specific antigen (ESA+)) was demonstrated to be a marker in colon CSCs [69]. However, additional studies showed that CD44+ cells reside throughout the entire crypt, including the proliferative compartment, suggesting that the CD44+ colon cells are not necessarily stem-like [65]. We have examined CD44 and ALDH together in one of our CRC patient-derived tumor xenograft (PDTX) models to determine if CD44+ cells had tumorigenic properties [70]. Despite ALDH+/CD44+ cells showing some tumorigenic growth, ALDH+/CD44− cells exhibited a higher incidence and faster growing tumors. In this same PDTX model, isolation and injection of ALDH+ and ALDH− cells in mice showed a significant difference with respect to tumor growth [70]. ALDH+ cells produced fast growing and large tumors when compared to ALDH− cells that either produced very small tumors or no tumors in five separate PDTX models. Importantly, all ALDH+ tumors looked morphologically the same as the original tumor. Several other studies have shown that injection of ALDH+ cells from colitis and colon cancer patients facilitated spheroid formation (in vitro three-dimensional spheroid cell culture that more closely resembles the in vivo environment) and tumor growth in a xenograft model, while ALDH− cells were incapable of tumor growth [31, 65]. These studies demonstrate that ALDH catalytic activity appears to be a robust marker of CSCs in CRC.
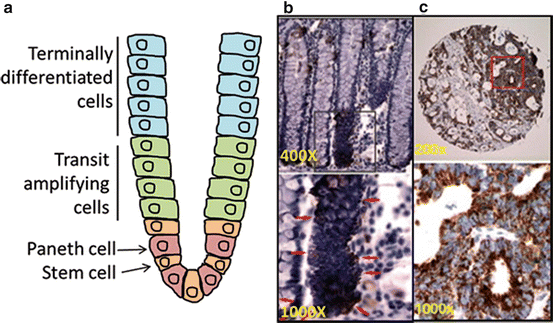
Fig. 16.3
ALDH1B1 expression pattern in normal colon and colon adenocarcinoma. Location of various cell types in normal colon (a). ALDH1B1 expression (red arrows) is strictly localized to stem-like cells at the base of crypts in the normal human colon (b). ALDH1B1 is expressed at extremely high levels throughout all cells of human colon adenocarcinomas (c). In figures (b, c) (reproduced from Chen et al. [71]), lower panels are higher magnification of areas identified by squares in the upper panel
Given the apparent promise of ALDH activity as a potential biomarker of CSCs, many investigations are currently exploring the role of ALDH in CSC function. In particular, a great deal of focus is being placed on which ALDH isoform(s) mediate the catalytic activity in the CSCs. In normal colon stem cells, ALDH1 has been demonstrated to be primarily expressed at the bottom of the crypt compartment in the colon (where colon-specific stem cells are located) and ALDH1 levels are significantly elevated in the development and progression of CRC [65]. Interestingly, ALDH1 protein levels are elevated in the colon of patients with ulcerative colitis (a risk factor for colon cancer) compared to normal colon cells; such expression may be important in the transformation from colitis to colon cancer [31]. We have shown that ALDH1B1 protein is 5.6-fold higher when compared to ALDH1A1 in CRC patients and may be a potential biomarker in CRC (Fig. 16.3b, c) [71]. Similarly, very high expression of ALDH1B1 was found in the colon polyps of Apc(Min)/+ mice (our unpublished data). While these studies indicate elevations in individual ALDH isoforms in CRC, the contribution of these enzymes to the progression of CSCs and CRC remain to be clarified.
A common problem associated with standard chemotherapeutic regimens in CRC is treatment resistance. Although chemotherapy is effective at reducing tumor burden, many CRC patients will experience disease recurrence and ultimately succumb to their disease. CSCs are thought to be responsible for chemotherapy resistance and disease recurrence [72]. Therefore, therapeutic elimination of this population would be predicted to reduce tumor recurrence and ultimately improve survival. In our CRC PDTX model, the effects of an inhibitor of the Notch pathway (considered to be important for self-renewal of colon stem cells) in combination with irinotecan was investigated on the ALDH+ cell population [70]. The combination therapy was effective at reducing the number of ALDH+ cells as well as tumor recurrence, even after treatment was discontinued when compared to single agent Notch pathway inhibition and irinotecan. Administration of the combination therapy for 28 days prevented tumor growth in the ALDH+ cell xenograft model; this protection continued for 3 months after combination treatment was completed [70]. These data indicate that the ALDH+ population has the ability to self-renew, and significantly reducing this population of cells delays tumor recurrence (Fig. 16.2). Whether specific ALDH isozymes contribute to chemotherapy resistance remains to be determined.
16.4.2 Pancreatic Cancer
Despite considerable research, the 5-year survival rate for pancreatic cancer still remains extremely poor. A concerted effort is underway to delineate pathways that are dysregulated in the CSC population of this disease and thereby identify novel potential therapeutic targets.
In pancreatic cancer, ALDH+ cells have been shown to possess stem cell features, as evidenced by enhanced clonogenicity in vitro and tumorigenic growth in mice [73]. These cells also have greater tumorigenic potential than CD133+ cells [73]. Interestingly, ALDH+ cells from pancreatic cancers have been demonstrated to: (1) express many genes of the mesenchymal phenotype, (2) have an increased capacity to migrate and invade, and (3) be more numerous in metastatic lesions [30]. Furthermore, in pancreatic cancer patients, expression of ALDH in tumors is associated with a worse survival rate than those tumors that do not express ALDH [30]. ALDH1A1 expression has been linked to resistance to chemotherapy in a pancreas PDTX model. In this context, treatment with gemcitabine was shown to enhance gene and protein expression of ALDH. Inclusion of an inhibitor of hedgehog (a pathway important for stem cell regulation in the pancreas) with gemcitabine resulted in decreased expression of ALDH [74]. These studies suggest that ALDH+ cells are stem-like cells in pancreas cancer and may be important contributors in disease progression and chemoresistance; therefore, contribute to the negative outcomes in patients with pancreatic cancer.
16.5 Summary
There is accumulating evidence that supports a role for ALDHs in cancer development and progression. The exact mechanisms by which ALDHs influence tumorigenesis remain to be defined. Certainly, metabolism of acetaldehyde and/or the generation of retinoic acid represent modalities by which ALDHs could influence CRC and pancreatic cancer. ALDH catalytic activity appears to be an excellent biomarker that can be utilized for the isolation and characterization of the CSC population in tumors obtained from patients with CRC or pancreatic cancer. It is becoming apparent that the various ALDH isozymes may have different roles in tumorigenesis (from metabolism of the carcinogen to modulation of the proliferation-regulating retinoids) and that the timing and cellular localization of isozyme expression may be critical factors that influence how ALDHs modulate cancer development and progression. Further studies are needed that identify (1) the importance of ALDH catalytic activity in modulation of tumorigenesis, (2) the specific ALDH isozymes involved (and that regulate CSCs), and (3) the signaling pathways that regulate tumor-associated ALDH expression. The results obtained from such studies should lead to the development of novel therapies that may more effectively treat these devastating diseases.
Related posts:
 A Perspective on Chemoprevention by Resveratrol in Head and Neck Squamous Cell Carcinoma
Transgenic Mouse Models for Alcohol Metabolism, Toxicity, and Cancer
A Perspective on Chemoprevention by Resveratrol in Head and Neck Squamous Cell Carcinoma
Transgenic Mouse Models for Alcohol Metabolism, Toxicity, and Cancer
 Genetic Polymorphisms of Alcohol Dehydrogense-1B and Aldehyde Dehydrogenase-2, Alcohol Flushing, Mean Corpuscular Volume, and Aerodigestive Tract Neoplasia in Japanese Drinkers
Genetic Polymorphisms of Alcohol Dehydrogense-1B and Aldehyde Dehydrogenase-2, Alcohol Flushing, Mean Corpuscular Volume, and Aerodigestive Tract Neoplasia in Japanese Drinkers
 Alcohol Consumption, Wnt/β-Catenin Signaling, and Hepatocarcinogenesis
Alcohol Consumption, Wnt/β-Catenin Signaling, and Hepatocarcinogenesis
 Alcoholic Cirrhosis and Hepatocellular Carcinoma
Alcoholic Cirrhosis and Hepatocellular Carcinoma
 Synergistic Toxic Interactions Between CYP2E1, LPS/TNFα, and JNK/p38 MAP Kinase and Their Implications in Alcohol-Induced Liver Injury
Synergistic Toxic Interactions Between CYP2E1, LPS/TNFα, and JNK/p38 MAP Kinase and Their Implications in Alcohol-Induced Liver Injury
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


