Abnormal Growth and Development
M. Joan Mansfield
Jonathan M. Swartz
KEY WORDS
Constitutional delay of puberty
Delayed puberty
Growth
Precocious puberty
Puberty
Short stature
This chapter focuses on the adolescent whose growth and/or development falls outside the range of normal. These issues are usually of enormous concern to adolescents and their family, and the health care provider must have a clear understanding of how to evaluate and manage these problems. In evaluating growth during adolescence, it is necessary to assess whether a teen has reached puberty, whether puberty is proceeding normally, and whether the bony epiphyses are still open to permit further growth.
Adolescents who are progressing normally through puberty may present with concerns about short stature. Most of these teens have genetic or familial short stature with other major categories including chronic disease, constitutional delay of growth and development, and endocrine diseases. Girls who are short may seek medical attention for this complaint when they have just reached menarche and worry that future growth in height will be limited. Boys may present as their pubertal growth spurt slows and they are still shorter than they had hoped. Most hormonal deficiencies, chronic diseases, and malabsorptive states that slow growth will also cause at least some delay in puberty or failure to progress normally through puberty; these are less likely causes for the short stature in teens who have normal puberty.
Definition of Short Stature
Adult height is strongly dictated by genetic factors; therefore, evaluation of short stature must be assessed considering the heights of family members. Generally, 2 standard deviation (SD) (2.3 percentile) below the mean height on a cross-sectional growth chart is used as the lower limit of normal.
Criteria for Evaluation An adolescent should be considered for an evaluation of short stature if:
Linear growth rate is <4 cm/year during the years prior to the normal age for peak linear growth velocity.
No evidence of a peak linear growth velocity by age 16 years in boys and 14 years in girls.
Deceleration below an individual’s established growth velocity occurs.
The adolescent’s height is more than 2 SDs below the calculated midparental height (see Chapter 2).
The adolescent’s height is more than 3 SDs below the mean. Consideration should be given to carrying out a full evaluation if an adolescent’s height is between 2 and 3 SDs below the mean; at a minimum, a careful history and physical examination, screening laboratory tests, and observation of growth for 6 months are warranted.
Differential Diagnosis
Familial short stature
Chronic illness—can include diseases such as cystic fibrosis, human immunodeficiency virus (HIV) infection, severe asthma, congestive heart failure, renal failure, inflammatory bowel disease, celiac disease, among others
Constitutional delay of puberty
Endocrine—can include hypothyroidism, isolated growth hormone (GH) deficiency, hypercortisolism, adrenal insufficiency, and poorly controlled diabetes mellitus
Congenital syndromes including Down syndrome (trisomy 21), Turner syndrome (45X), Noonan, Hurler, Silver-Russell syndrome, Laron syndrome (GH receptor gene mutations), short stature homeobox gene (SHOX) deficiency
Intrauterine growth retardation or small for gestational age
Skeletal dysplasias—including chondrodysplasias (often have abnormally short extremities)
Evaluation
History
Maternal pregnancy history—medical illnesses and medication use
Birth weight and length, and estimate of gestational age—important because premature infants with appropriate small weight tend to have a normal growth potential, whereas infants with intrauterine growth retardation who are inappropriately small for gestational age may not have catch-up growth
Complete review of systems:
Renal—polyuria and polydipsia for hypothalamic and/or pituitary disorders
Cardiac—peripheral edema, murmurs, and cyanosis
Gastrointestinal—diarrhea, flatulence (malabsorption), vomiting, and/or abdominal pain
Pulmonary—sleep apnea, asthma, or symptoms suggestive of cystic fibrosis
Neurological—headaches, visual field defects suggesting pituitary neoplasms
Growth history—review of symptoms and growth charts
Family history—adult height, and patterns of growth and puberty in first- and second-degree relatives
Dietary history
Physical Examination
A complete physical examination is the next step in the evaluation and should include:
Height and weight (including growth measurements that are plotted on appropriate developmental charts for height, weight, and body mass index [BMI])
Arm span and upper-to-lower (U/L) body-segment ratio
Sexual maturity ratings (SMRs)—specifically breast and genital development, and not pubic hair only
A general physical examination, with special attention to the thyroid gland, ophthalmological examination, neurological examination, and stigmata of congenital syndromes
Laboratory/Radiologic Evaluation
The laboratory evaluation of short stature should include the following:
Routine laboratory screening: Includes complete blood cell (CBC) count with differential, sedimentation rate, urinalysis (under age 3), chemistry profile including serum creatinine, insulin-like growth factor-1 (IGF-1), and thyroid-stimulating hormone (TSH) and free thyroxine (free T4). Screening for celiac disease with total immunoglobulin A and anti-tissue transglutaminase is also advised in asymptomatic short children.
Bone age: X-ray of the left hand and wrist for bone age (since the bone age can determine if there is more potential for growth and be used to estimate predicted final height).1
Midparental height calculation: It is also useful to obtain the parents’ heights and calculate a midparental height (formula provided in Chapter 2). Although there are many genes involved in stature, and an offspring’s height frequently varies considerably from midparental height, the midparental or target height can still give a good clue that the short stature is genetic.
Karyotype: A karyotype is useful when signs of Turner syndrome are present, as well as in girls with unexplained short stature and otherwise normal labs. It can also be useful in boys with genital anomalies.
Other tests: Other tests may be indicated depending on the history and physical examination and may include:
Central imaging studies—cranial magnetic resonance imaging (MRI) with contrast
Gastrointestinal studies
Endocrine studies:
Serum levels of IGF-1 and insulin-like growth factor-binding protein 3 (IGFBP-3) are often assessed when the growth pattern is concerning for GH deficiency. IGFBP-3 can be useful in underweight or nutritionally deficient children who have low IGF-1 levels.
GH stimulation testing is usually done by a pediatric endocrinologist using one of several protocols (insulin, glucagon, arginine, L-dopa, or clonidine). Two tests are usually carried out together and the patient is considered GH deficient if the GH response is <7 to 10 ng/mL on both tests. In prepubertal adolescents, priming with estrogen (in males and females) before stimulation testing should be considered for maximal GH release.
Genetic testing: SHOX testing may be considered when a constellation of typical features are present such as short stature, increased upper/lower segment ratio, reduced arm span-to-forearm length ratio, cubitus valgus, Madelung wrist deformity, forearm bowing, short metacarpals or metatarsals, apparent muscular hypertrophy, increased BMI, high arched palate, abnormal auricular development, micrognathia, or short neck.2
Suggestions for Diagnosis
Constitutional delay of puberty: Most short stature in adolescents is the result of either constitutional delay of puberty or familial short stature. Guidelines for diagnosis are outlined later in this chapter.
Genetic or familial short stature: Genetic or familial short stature is suggested by the following:
Normal history and physical examination findings
Family history of short stature
Growth curve that generally parallels the 3rd percentile
Bone age that is appropriate for chronological age
Chronic illness: Chronic renal disease and Crohn’s disease are additional causes of short stature. These diseases are usually diagnosed by an abnormal history, physical examination findings, or results of tests including screening CBC, sedimentation rate, urinalysis, and chemistry studies. Renal tubular acidosis can be overlooked as a cause of short stature, especially in children under 3. This process may be suggested by family history, urine pH level, or serum bicarbonate values.
Endocrine causes: Endocrine causes of short stature, such as hypothyroidism, GH deficiency, adrenal insufficiency, and adrenocortical excess, are less common. Hypothyroidism and adrenocortical excess can usually be detected by the patient’s history, physical examination including a growth chart, or screening laboratory tests. Adolescents with classic GH deficiency can be difficult to differentiate from those with constitutional delay of puberty. This is particularly difficult during the time of expected peak linear growth velocity, when the growth of an adolescent with constitutional delay of puberty slows from the normal growth curve as other adolescents accelerate their growth velocities. Individuals with classic GH deficiency have normal body proportions and often a high-pitched voice, a tendency toward hypoglycemia, a microphallus in boys, a child-like face, soft and finely wrinkled skin, and a large prominent forehead.
Treatment of Short Stature with GH
GH Deficiency
Patients with classic GH deficiency have marked benefit in statural outcome as the result of GH treatment. In addition, those with complete GH deficiency benefit from treatment (from the metabolic effects of GH) with regard to improving bone density, decreasing fat mass, and improving muscle strength, even if epiphyseal fusion has been achieved. It appears that these subjects should continue GH treatment at a markedly reduced dose, compared with that used for growth augmentation, throughout life.
Bioengineered human GH has been available since the 1980s. Patients with classical GH deficiency usually present with extreme short stature and slow growth (<4 cm/year) well before adolescence, although acquired GH deficiency, sometimes due to head trauma or tumors, may present in adolescence with slow growth and relatively delayed puberty.3
Other Conditions
Turner Syndrome: GH has been used to increase height velocity and increase final adult height in patients who do not have GH deficiency by GH stimulation tests. GH is approved for use in patients with short stature due to Turner syndrome using a higher dose than is recommended for GH deficiency (0.05 mg/kg/day subcutaneously or 0.35 mg/kg/week for Turner syndrome). IGF-1, thyroid screens, and bone ages by x-ray are monitored during therapy. Patients with Turner syndrome should have baseline renal ultrasonography and periodic cardiac MRI and echocardiograms to screen for aortic root enlargement. Aortic dissection is a rare but potentially fatal cause of severe chest pain in patients with Turner syndrome. GH treatment should ideally be initiated early in childhood when growth rate begins to fall off. Estrogen replacement is usually delayed to age 12 to 14 or sometimes later to maximize height gain in patients with Turner syndrome. GH can also be used similarly in Noonan syndrome.
Intrauterine Growth Retardation: GH is also approved by the US Food and Drug Administration (FDA) for use in patients with short stature due to intrauterine growth retardation without catch-up growth, Prader-Willi syndrome, and chronic renal failure before transplantation. GH has also been approved for treatment of children and adolescents with idiopathic short stature who are more than 2.25 SD below the mean in height and who are unlikely to catch up in height.4,5,6,7 Patients who qualify for a trial of treatment with human GH for idiopathic short stature must have open epiphyses permitting further height gain. Patients with severe short stature who desire treatment with GH should be referred to a pediatric endocrinologist.
Definition
In general, 2 SDs above and below the mean are used to define the range of normal variability. Chapter 2 is helpful in determining guidelines for evaluation, and further guidelines are discussed subsequently.
Delayed development is defined by the absence of breast budding by age 13 in girls or the lack of testicular enlargement by age 14 in boys, both 2.5 SD beyond the average age at onset of these changes. Alterations in the chronological relationship of pubertal events are also common causes for evaluation. These include phallic enlargement in the absence of testicular enlargement in boys or the absence of menarche by age 16, or 4 years after the onset of breast development, in girls. If puberty is interrupted, there is a regression or failure to progress in the development of secondary sexual characteristics, accompanied by a slowing in growth rate.
General Guidelines for Evaluating Puberty
In Males
A male adolescent may be considered to have delayed puberty if:
Genital stage 1 persists beyond the age of 13.7 years, or pubic hair stage 1 (PH1) persists beyond the age of 15.1 years.
More than 5 years have elapsed from initiation to completion of genital growth.
In Females
A female adolescent may be considered to have delayed maturation if:
Breast stage 1 persists beyond the age of 13.4 years, PH1 persists beyond the age of 14.1 years, or there is failure to menstruate beyond the age of 16 years.
More than 5 years have elapsed between initiation of breast growth and menarche.
These general guidelines must be considered in the context of the teen’s family history as to growth and pubertal development, his or her previous growth pattern, and with regard to the review of systems and physical examination.
Differential Diagnosis
Delayed development occurs more commonly in boys than in girls. Most patients who present for an evaluation of slow growth and delayed development are high school-aged boys who are concerned about their short stature, as well as their lack of muscular and secondary sexual development, which puts them at a disadvantage among their peers. Most of these boys have constitutionally delayed development; however, the clinical presentation of the patient with constitutional delay may be indistinguishable from that of the patient whose pubertal delay is the result of an organic lesion.
Constitutional Delay of Puberty
Adolescents with constitutional delay of puberty have often been slow growers throughout childhood. In the absence of sex steroids of puberty, growth may slow even further to <5 cm/year as these children reach an age when puberty would normally occur. Growth velocity increases into the normal range when these teens finally enter puberty. Adolescents with constitutional delay of puberty often have a family history of delayed growth and development in relatives. Teens with constitutional delay of puberty eventually enter puberty on their own. Although they have a longer time to grow before their epiphyses close, they tend to have a less exuberant growth spurt than earlier developers so that their final height is often shorter than average.
Functional Causes of Delayed Puberty
Gonadotropin-releasing hormone (GnRH) secretion can be inhibited centrally by the following:
Inadequate nutrition, including eating disorders
Chronic disease including chronic heart disease, severe asthma, inflammatory bowel disease, celiac disease, rheumatoid arthritis, chronic renal failure, renal tubular acidosis, sickle cell anemia, diabetes mellitus, systemic lupus erythematosus, cystic fibrosis, and infection with HIV
Severe environmental stress
Intensive athletic training
Hypothyroidism and excess cortisol states
Drugs such as opiates and stimulants
Eating disorders associated with self-imposed restriction of caloric intake can delay or interrupt the progression of puberty. Anorexia nervosa most often develops in girls in early to middle adolescence, who have already entered puberty. Young adolescent boys or girls who are dieting because of fear of obesity may present with the complaint of delayed development. Crohn’s disease or celiac disease may also present with delayed development and poor growth as the major symptoms. Since adolescence is normally a period of rapid growth and weight gain, failure to gain or small amounts of weight loss may be manifestations of significant nutritional insufficiency. Poor growth and delayed puberty are common in cystic fibrosis, thalassemia major, renal tubular acidosis, renal failure, cyanotic congenital heart disease, sickle cell anemia, systemic lupus erythematosus, acquired immune deficiency syndrome, or very poorly controlled asthma or type 1 diabetes mellitus. Patients who are on stimulants such as methylphenidate (Ritalin) for treatment of attention deficit disorder may have decreased appetite because of the medication and slower growth rates as a result of nutritional insufficiency.
Hypothyroidism may present in an adolescent with slowing of height velocity (height-dropping percentiles on the growth chart) whose weight is well preserved for height or who is mildly overweight, sometimes with delayed or interrupted pubertal development. The classic signs include dull dry skin, perhaps with scalp hair loss, decrease in pulse rate and blood pressure, constipation, and cold intolerance. A goiter is not always present. Autoimmune thyroiditis is the most common cause of hypothyroidism in teens. There may be a family history of hypothyroidism or autoimmune issues.
Cushing syndrome (endogenous glucocorticoid overproduction) or chronic exposure to high doses of glucocorticoids for medical treatment causes excessive weight gain, slowing of height velocity, and may interrupt or delay puberty or, if endogenous sex steroid production is also increased, may present with precocious puberty without a growth spurt (Fig. 11.1).
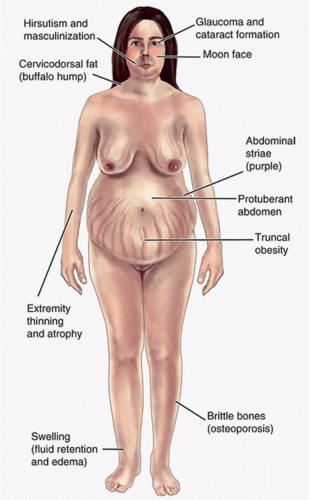 FIGURE 11.1 Typical features consistent with Cushing syndrome including central and cervical adiposity, moon face, thin extremities, along with violaceous striae. Hirsutism can be noted in females. (From Diane SA, Samantha JV. Drug therapy in nursing. 4th ed. Philadelphia, PA: Lippincott Williams & Wilkins, 2011.) |
Hypothalamic Causes of Delayed or Absent Puberty
The ability of the hypothalamus to secrete GnRH may be damaged by:
Local tumors (germinomas, craniopharyngiomas, astrocytomas, or gliomas)
Infiltrative lesions such as central nervous system (CNS) leukemia or histiocytosis X
CNS irradiation
Traumatic gliosis
Mass lesions such as brain abscesses or granulomas due to sarcoidosis or tuberculosis
Congenital defects in the ability to secrete GnRH (including isolated hypogonadotropic hypogonadism and Kallman syndrome). Recent studies have confirmed a number of genes involved in cases of hypogonadotropic hypogonadism including but is not limited to GNRHR, KISS1R, TAC3, TACR3, GnRH1, KAL1, FGFR1, PROK2, PROKR2, FGF8, CHD7, WDR11, and NELF.8
Congenital brain malformations associated with inability to secrete GnRH include septooptic dysplasia.
Pituitary Causes of Delayed Puberty
Puberty may not begin or may fail to proceed if the pituitary cannot respond to GnRH stimulation with luteinizing hormone (LH) and follicle-stimulating hormone (FSH) production. This may be due to:
Pituitary tumor
Selective impairment of gonadotrope function by hemochromatosis
Congenital hypopituitarism, which is usually diagnosed either in the neonatal period or with poor growth during childhood; causes include genetic defects that interfere with pituitary formation and empty sella syndrome.
Acquired hypopituitarism
Prolactinoma—excessive prolactin production by a pituitary adenoma (prolactinoma) or other tumor may interrupt or prevent puberty by interfering with gonadotropin production. Patients with prolactinomas most often present with secondary amenorrhea often with galactorrhea, but may present with stalled puberty. Headaches are sometimes present. Prolactinomas are more common in girls than boys, but can occur in both. Psychotropic drugs such as antipsychotics are a frequent cause of hyperprolactinemia.
Gonadal Failure
If the gonads are unable to respond to LH and FSH, puberty will not proceed.
The causes of gonadal failure with abnormal karyotype include:
Gonadal dysgenesis: The most common cause of gonadal failure is gonadal dysgenesis, which occurs in association with abnormalities of sex chromosomes. The gonads fail to develop and become rudimentary streaks. These patients are phenotypic females with normal immature female genitalia. The most common phenotype is Turner syndrome, which is caused by absence of part or all of a second sex chromosome. These patients are typically short with a final untreated height averaging 143 cm. Other identifying features of Turner syndrome are low-set ears, a webbed neck, widely spaced nipples, a trident hairline, an increased carrying angle of the lower arms, and short fourth and fifth fingers and toes. Renal abnormalities such as duplications and horseshoe kidney, and left-sided cardiovascular abnormalities such as bicuspid aortic valve, dilatation of the aortic root and coarctation of the aorta are also associated with Turner syndrome. Half of these patients have 45, X karyotypes, whereas the rest are mosaics or have various X chromosome abnormalities or deletions.
Klinefelter syndrome: Males with Klinefelter syndrome (47, XXY) may present with poorly progressing puberty caused by partial gonadal failure. Their testes can make some testosterone when driven by high levels of gonadotropins (LH and FSH) and they tend to have significantly impaired sperm production.9 In the 47, XXY patient with pubertal development, the testes become small and firm because they become fibrotic. Gynecomastia and eunuchoid body habitus are often seen.
The causes of gonadal failure with normal karyotype include:
Acquired gonadal disorders
Infection—viral or tubercular orchitis or oopheritis
Trauma—bilateral testicular torsion resulting in anorchia is another cause of gonadal failure in malesRelated posts:

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree