Introduction
Modern medicine has managed to increase human lifespan by finding better treatments for diseases that at one time were incurable. Diseases like smallpox, carried by the variola virus, killed thousands of people yearly before vaccines were discovered . Not to mention the thousands of lives saved with annual influenza vaccines, or the millions who died worldwide from the Spanish Flu of 1918–19, as well as the current COVID-19 pandemic and the effect of vaccines there . Furthermore, thousands died every year from bacterial infections caused by Escherichia coli and Klebsiella pneumoniae until Alexander Fleming discovered antibiotics in 1928 . In contrast, the increase in lifespan resulting from medical breakthroughs has not been accompanied by a concomitant rise in health-span, which is the period an individual is generally healthy and free of chronic diseases. The vast majority of diseases worldwide are chronic diseases that have age as a primary, if not the leading underlying cause. Moreover, according to the Center for Disease Control and Prevention (CDC), chronic diseases cost 3.5 trillion dollars to the healthcare system annually . Furthermore, it is estimated that 90% of people aged 65 or older have at least one chronic disease, while 75% have a minimum of two comorbidities . As such, there is imperative to find better treatments against chronic diseases that affect health and everyday activities, especially those whose underlying mechanisms may be shared; this includes osteoporosis and sarcopenia. Chronic diseases currently limit the human health-span to about 65 years, whereas a healthy human lifespan falls around 72 years. Age is the primary underlying risk for chronic degenerative diseases like osteosarcopenia; hence, novel and more effective ways to target the aging process are necessary . Over the past few years, one of the primary mechanisms integrating and driving aging in cells and tissues has been identified, cellular senescence. Consequently, the development of treatments that can destroy senescent cells, or block their spreading dysfunction to neighboring cells, such as inhibiting their amplification via Senescence Associated Secretory Phenotype (SASP) molecules, is desirable. The SASP involves the expression of multiple molecular processes that result in the secretion of proinflammatory products and extracellular matrix modifiers, including cytokines, growth factors, and proteases . These secreted proteins can ultimately induce nonsenescent cells to undergo senescence, increasing the percentage of senescence cells in a tissue to as high as 20% of the cell population .
Thus, the approach of targeting the SASP would also potentially reduce or delay the senescent cell population growth or alleviate some of the senescence’s many deleterious effects. For a chronic disease like osteoporosis, senescence would include dysfunctional SASP generating osteoprogenitor cells, osteoblasts, and osteocytes resulting in decreased bone formation, which would be delayed, reduced, or reversed if effective treatments were available, thus increasing patient health-span in the process. In the sections that follow, we will be discussing three major areas of ongoing research that show promise for decreasing the burden of senescent cells on bone and muscle health. We will start by discussing the dual role of autophagy on cellular senescence, highlighting its pro- and antisenescence capabilities. We will discuss senotherapeutic agents, namely senostatics and senolytics. These are novel classes of agents with the potential of treating chronic diseases like osteoporosis and sarcopenia by either inhibiting the SASP produced by senescent cells blocking the spread of senescence, restoring cell function, or inducing senescent cell apoptosis, eliminating senescent populations from tissues broadly. Finally, we will discuss the advantages and disadvantages of each senotherapeutic approach, as well as some of the current limitations on the field, for example, the lack of precise and accurate biomarkers that will be required to successfully implement senotheraputics in the treatment of chronic disease.
Senescence aging effects on bone and osteoporosis
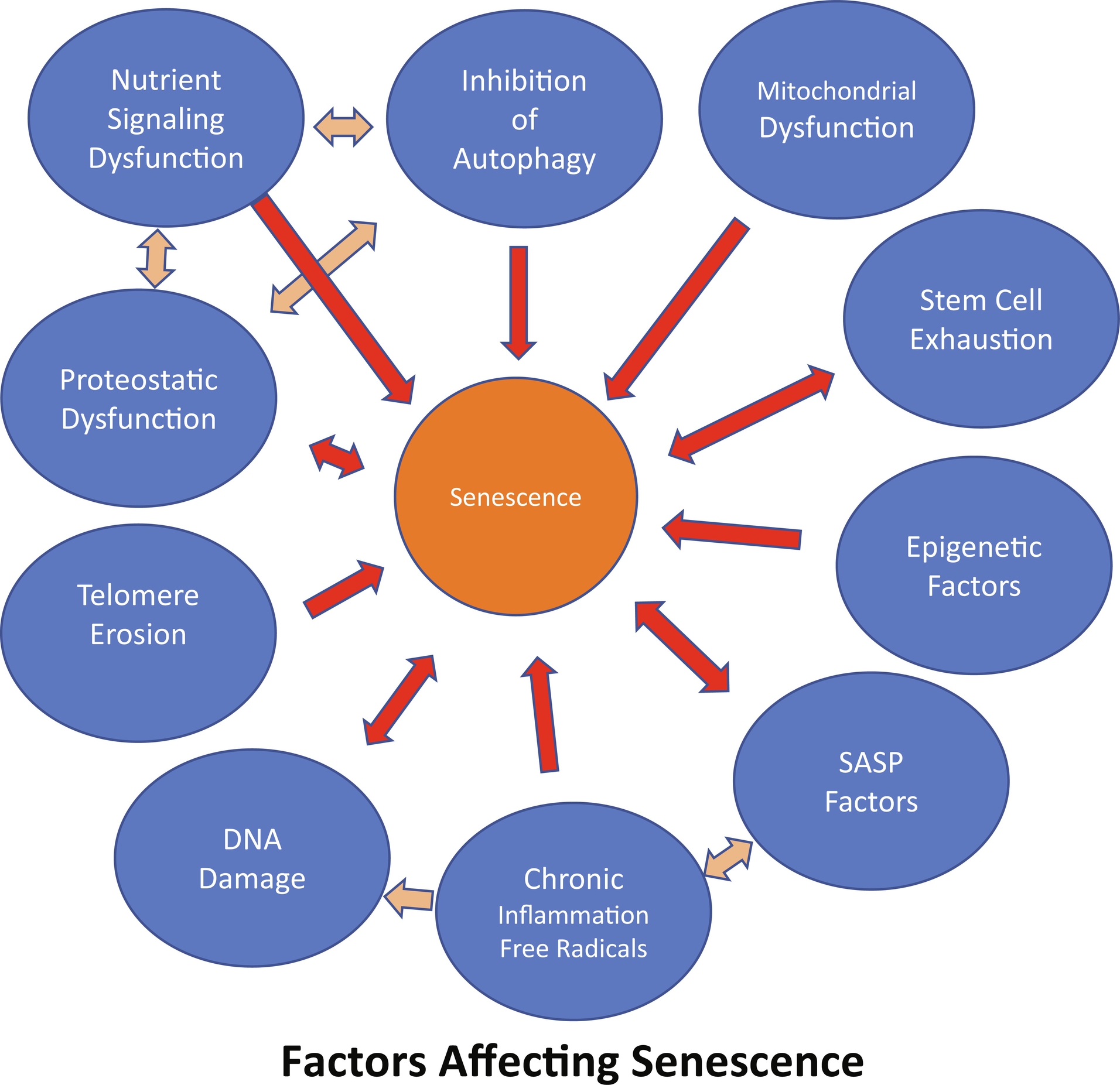
Senescence is a process by which a cell enters growth arrest and stops dividing. This usually happens when the cell is exposed to different types of cellular stress like oxidative, genotoxic, or oncogenic ( Fig. 1 ). Furthermore, “replicative” senescence can also happen when the telomeres become shortened to a point where they are no longer stable . Evolutionarily senescence is an early process; hence, it must serve a vital positive role in maintaining homeostasis at the cellular and organism levels. Indeed, there is abundant evidence of the beneficial roles of cellular senescence. Among them, research has shown it is essential in suppressing neoplastic growth and in wound healing . Nonetheless, cellular senescence can pose detrimental health effects when the senescent cell populations increase to levels that interfere with normal cell and tissue function. The quantity of senescent cells in the body is mainly kept under control by the host’s immune system. However, there is growing evidence that the immune system becomes dysfunctional with age. This reduction in immune surveillance/function allows senescent cells to increase and accumulate tying into, and helping to drive, most chronic diseases, including osteoporosis .
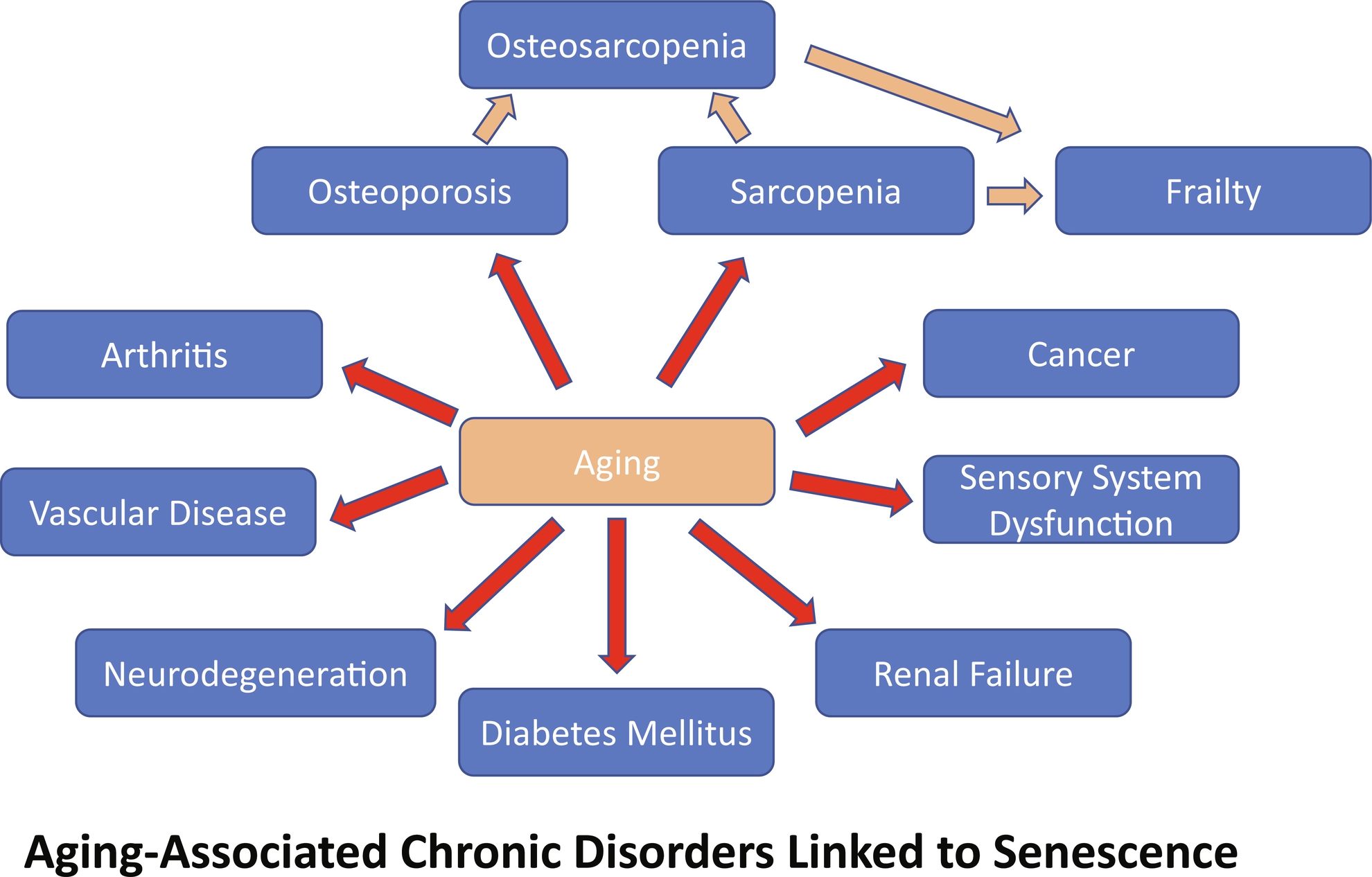
It is now widely accepted that the underlying cause of many chronic diseases like osteoporosis and sarcopenia, which tend to appear with advancing age, is age itself or processes linked to aging ( Fig. 2 ). Furthermore, evidence suggests that senescent cells tend to increase exponentially with advancing age, suggesting a link between cellular senescence and aging. Senescent cells are characterized by developing a SASP. This phenotype is responsible for chronically producing a myriad of proinflammatory products. Among these, cytokines like IL-15 and IL-1β, growth factors such as Insulin-like Growth Factor Binding Protein-5 (IGFBP-5), and proteases like matrix metalloproteinases (MMP1 and MMP3) . The host’s immune system usually clears out these SASP products, thereby maintaining homeostasis. The problem arises when the organism undergoes aging. In mice, this happens after 18 months of age, whereas humans typically start entering the senior age after 65 years of age . Thus, by 18 months of age, a mouse is similar to a 65 +-year-old human. At this point in the aging process, the immune system becomes less functional, making it difficult for the organism to eliminate the SASP proinflammatory products. When these agents are not adequately removed from tissues, they contribute to chronic inflammation, which eventually contributes to multiple diseases, including degenerative diseases, osteosarcopenia being only one of them. Aging, unfortunately, takes its toll in a multiorgan system manner, affecting the immune system, cardiovascular system, metabolome, nervous system, and musculoskeletal system ( Fig. 2 ). In bone, aging eventually compromises osteocytes, with a subset of these bone cells, as well as earlier differentiating and even progenitor populations becoming senescent and developing a SASP releasing proinflammatory products that act in a paracrine or endocrine manner to produce local and systemic inflammation .
Altogether, in bone, increased osteocyte senescence and SASP products in the local environment and systemic circulation disrupt bone homeostasis by inhibiting osteogenesis and promoting adipogenesis of bone marrow mesenchymal stem cells (BMSCs). This change in the lineage commitment of MSCs toward adipogenesis instead of osteogenesis and inhibition of osteogenesis results in net bone loss by disrupting the balance between bone formation and bone resorption. Ultimately, these age-related, senescence-based alterations in bone homeostasis can advance to the degree of degenerative bone diseases such as osteoporosis and osteosarcopenia.
Unfortunately, osteoporosis is becoming rampant with increased lifespans, with menopause for women and aging for both men and women being primary factors. Over 200 million people currently have osteoporosis. In addition, it is estimated that 1 in every 3 women over the age of 50, and 1 in every 5 men over the age of 50 will suffer from osteoporotic fractures in their lifetime . Furthermore, it is estimated that 25% of patients who suffer an osteoporotic hip fracture will die within a year of the event . Osteoporotic fractures are also associated with sarcopenia and falls. Considering the health costs and morbidities resulting from inadequate bone and muscle health with the newer understanding of the role of senescence driving this process, it highlights the need for research to understand the role of senescence in aging for bone loss as well as more broadly in chronic age-associated disorders. Research studies to date emphasize the need to develop treatments that can either kill senescent cells or inhibit their production of SASP proinflammatory and prosenescent products. More importantly, the development of more accurate and precise biomarkers for detecting senescent cells is imperative. Currently, it requires a number of biomarkers employed together in order to identify senescent cells, since individually they lack precision and accuracy in demonstrating senescence. Furthermore, different cells and tissues have different profiles of SASP factors and other biomarkers. Common biomarkers currently used to identify senescent cells in vitro and in vivo include: increased cell size, intracellular protein content, increased expression of p16, p21 and different combinations of SASP factors (like IL-6, IL-1α, IL-8) depending on cell or tissue type, increased cellular senescence-associated β-galactosidase (SA-βgal) activity, cytoplasmic HMGB1, and the presence of senescence-associated distension satellites (SASDS) . These biomarkers can identify various parameters which tend to be present in senescent cells. For example, the overexpression of the tumor suppression genes p16 and p21, give senescent cells the ability to bypass apoptosis. The β-galactosidase activity is increased in senescent cells, originates from the lysosome, and can be selectively detected at a suboptimal pH of 6.0 in senescent cells . Although β-gal activity is primarily in senescent cells, its activity is not required for senescence. Its elevation may be simply due to increased lysosomal populations in senescent cells.
A role for autophagy in antiaging
Autophagy is best known as a survival mechanism employed by a cell under multiple stressors, classically in low nutrient or oxygen conditions (for useful autophagy reviews, see Kirkin, 2020; Klionsky et al., 2016) . In response to nutrient deprivation, i.e., amino acid starvation, the cell induces autophagy which is a catabolic process during which the autodegeneration of specific organelles and macromolecules occurs in order to generate needed nutrients and building blocks for essential macromolecules via cellular recycling. Importantly, autophagic catabolism serves to eliminate unwanted and harmful elements such as misfolded proteins and damaged organelles; critically, this includes aged or damaged mitochondria (mitophagy) . The elimination of mitochondria by autophagic catabolism is a type of selective autophagy termed mitophagy. There are many examples of selective autophagy during which specific organelles, proteins, and intracellular pathogens are degraded, with a recent review of the multiple selective autophagy pathways and the key roles of the selective autophagy receptors like ATG19 and p62/SQSTM1, which are used to move targeted molecules or organelles into autophagosomes . Mitophagy, in particular, has been linked to cellular senescence by elegant studies utilizing pharmacological and genetic approaches that have been recently reviewed . They show Senescence-Associated Mitochondrial Dysfunction (SAMD) appears to be necessary for at least part of the specific SASP and may be responsible for tissue-level metabolic dysfunction associated with aging. In these studies, impaired mitophagy promotes senescence, which is consistent with the fact that mitochondrial dysfunction is an independent hallmark of aging .
As with other types of selective autophagy, mitophagy utilizes the core autophagy machinery . The core machinery of autophagy is dependent on many genes, some of which are designated as autophagy-related genes or Atg . To date, there are at least 30 Atg genes, and many of these genes encode products (ATG proteins) required for the formation of the autophagosome, the functional unit of autophagy. The autophagosome is a cytoplasmic organelle enclosed by a double membrane that must fuse with the lysosome for the contents within the autophagosome to be degraded. The fusion of the autophagosome with the lysosome generates an organelle referred to as the autolysosome, within which hydrolytic enzymes degrade the macromolecules that were initially sequestered by the autophagosome. There are at least nine genes involved in autophagy initiation, many of which regulate phagophore nucleation. The phagophore is the initial membrane structure that ultimately elongates and develops into the autophagosome, which is enclosed by two membranes, with the outer membrane eventually fusing with the lysosome membrane to form the autolysosome. A minimum of seven genes is involved in phagophore expansion. These genes and their specific functions have been recently reviewed, along with genes involved in the complex process of cargo sequestration within the autophagosome . Induction of autophagy often consists of inhibiting the mammalian target of Rapamycin or mTOR, a master cell growth regulator, a serine/threonine kinase. The regulation of mTOR action is quite complex and involves at least two distinct protein complexes that dictate mTOR action, with blockade of the mTOR complex 1 (mTORC1) able to induce autophagy. The complex mTORC1 includes ATG13, which is released from the complex in response to amino acid starvation such that it can function in autophagy initiation. The targeting (or downregulating) of specific Atg genes or the inhibition of mTOR have been common approaches to establish a role for autophagy in senescence.
A dual role for autophagy in mesenchymal stem cells: Antisenescence versus prosenescence
Although many of the early studies addressing a role for autophagy in senescence have been conducted in fibroblasts, recent studies suggest a role of autophagy in regulating cellular senescence of mesenchymal stem cells (MSCs). The functional roles of autophagy in MSCs, however, are not entirely clear. Evidence supports autophagy as a prosenescence process, albeit evidence also exists that derogates a prosenescence role for autophagy and further suggests that autophagy prevents cellular senescence . Concerning the antisenescence role of autophagy, recent studies showed that inducing elevations in autophagy in aged mice increased health-span by restoring stem cell activity . Other studies showed that autophagy is not only essential for sustaining the differentiation of stem cells, but that without autophagic activity, MSCs senescence is almost certain . For example, research done on primary human fibroblasts demonstrated that inhibiting autophagy, by either knocking down (via shRNA targeting) the Atg7 or Atg5 genes, prompted the fibroblasts to enter a senescent state, due to the accumulation of reactive oxygen species (ROS) and mitochondrial impairment . Indeed, depending on the cell/tissue and context, autophagy can inhibit or induce senescence with selective autophagy inhibiting senescence via depletion of key transcription factors and general autophagy, allowing the recycling of amino acids in stressed cells to form SASP molecules . These types of genetic studies provided strong support for an earlier study that established a key role for autophagy as an antiaging process in aged mice . This in vivo study showed increased lifespan if mice were treated with Rapamycin, a drug that blocks mTOR activity and induces autophagy. In addition, treating MSCs with Rapamycin resulted in increased autophagy and reduced senescence, whereas reducing Rapamycin resulted in decreased autophagy and increased senescence . We have recently confirmed this inverse relationship between autophagy inhibition and senescence induction in MSCs treated with kynurenine, an age related tryptophan metabolite, signaling via the aryl hydrocarbon receptor system leading to osteogenic inhibition . Moreover, investigations also have found that with aging, MSCs autophagic activity diminishes relative to MSCs from younger sources . This purported antisenescence role of autophagy on MSCs is intriguing and establishes a link between aging in bone, autophagy, and cellular senescence. This suggests that autophagy, at least partly, may be essential in maintaining homeostasis between bone deposition and bone resorption .
In human MSCs, one of the most used and reliable biomarkers to detect cells undergoing autophagy is LC3-II, specifically the PE lipidation of LC3-I to LC3-II. LC3 is the product of Atg 8, is typically upregulated during autophagy induction, and is required to form the autophagosomal membrane. Surprisingly, this biomarker has been found in cells entering replicative senescence, suggesting that autophagy plays a role in either promoting or maintaining replicative senescence . A more focused investigation conducted in MSCs showed that cellular senescence was dependent on autophagy induction. In this study, the data showed that when insulin-like growth factor 1 (IGF1) was diminished, the senescent cells were protected by autophagic activity even under hypoxic conditions . A similar prosurvival role for autophagy was reported in an earlier study that showed high glucose concentrations could trigger MSC cellular senescence by promoting the formation of reactive oxygen species (ROS), which then upregulates autophagic activity . MSCs cultivated at high glucose concentrations exhibited telomere attrition and genomic instability, and a higher rate of autophagic activity was required for their survival. This was assessed by measuring the quantity of various biomarkers, including Beclin-1 (ATG6), ATG5, ATG7 and LC3-II (ATG8). The biomarkers were found to be at higher than basal levels, suggesting a higher rate of autophagic activity and thus, a link between autophagy and MSCs senescence . Also, studies of the tumor suppressor p53 known to be involved in replicative senescence in mesenchymal stromal cells, show that p53 also upregulates autophagic activity , providing a molecular link between autophagy and replicative senescence . Another study focused on a type of cellular senescence called Oncogene-Induced Senescence (OIS). Senescent cells induced by oncogenic stress exhibit proliferation arrest with metabolic reprogramming as well as senescence-associated inflammation. This research suggested that in OIS fibroblast cells, autophagy was upregulated. Still moreover, autophagy was regulating the SASP by providing the biochemical building blocks needed to synthesize the inflammatory proteins and cytokines for secretion . All this evidence suggests that autophagy, probably general autophagy versus selective autophagy, supports cellular senescence under certain circumstances. Thus, autophagy has a dual role in regulating senescence in MSCs and likely in bone and muscle homeostasis. Autophagy as an antisenescence process is predicted to help promote bone and muscle growth. Still, autophagy could also aid senescent-based bone and muscle loss as a prosenescence facilitator.
Reconciliation regarding autophagy and senescence
While there is evidence supporting a dual role of autophagy in cellular senescence, the molecular details of autophagy-mediated regulation of pro- versus antisenescence are limited. The current standing regarding the intricate relationship between autophagy and senescence is that it depends on whether a cell undergoes general or selective autophagy and on the triggering stimulus. Evidence suggests that under certain stimuli, i.e., d -galactose and oxidative stress, or mitophagy, selective autophagy has antisenescence capabilities . Whereas under stimuli-like glucose and hypoxia, general autophagy showed prosenescence capabilities . However, autophagy induced by replicative exhaustion has been linked to both anti- and prosenescence. The fact that replicative exhaustion as an autophagy stimulus has been found to drive senescence and reduce senescence underscores the need for continued research to molecularly define the effects of replicative exhaustion on autophagy and senescence. Currently, we believe that the timing of induction of general autophagy also plays an important role in determining if replicative exhaustion promotes or reduces senescence. When general autophagy acts early to maintain homeostasis, it exhibits antisenescence properties. In contrast, when general autophagy acts late to maintain homeostasis, it shows prosenescence properties .
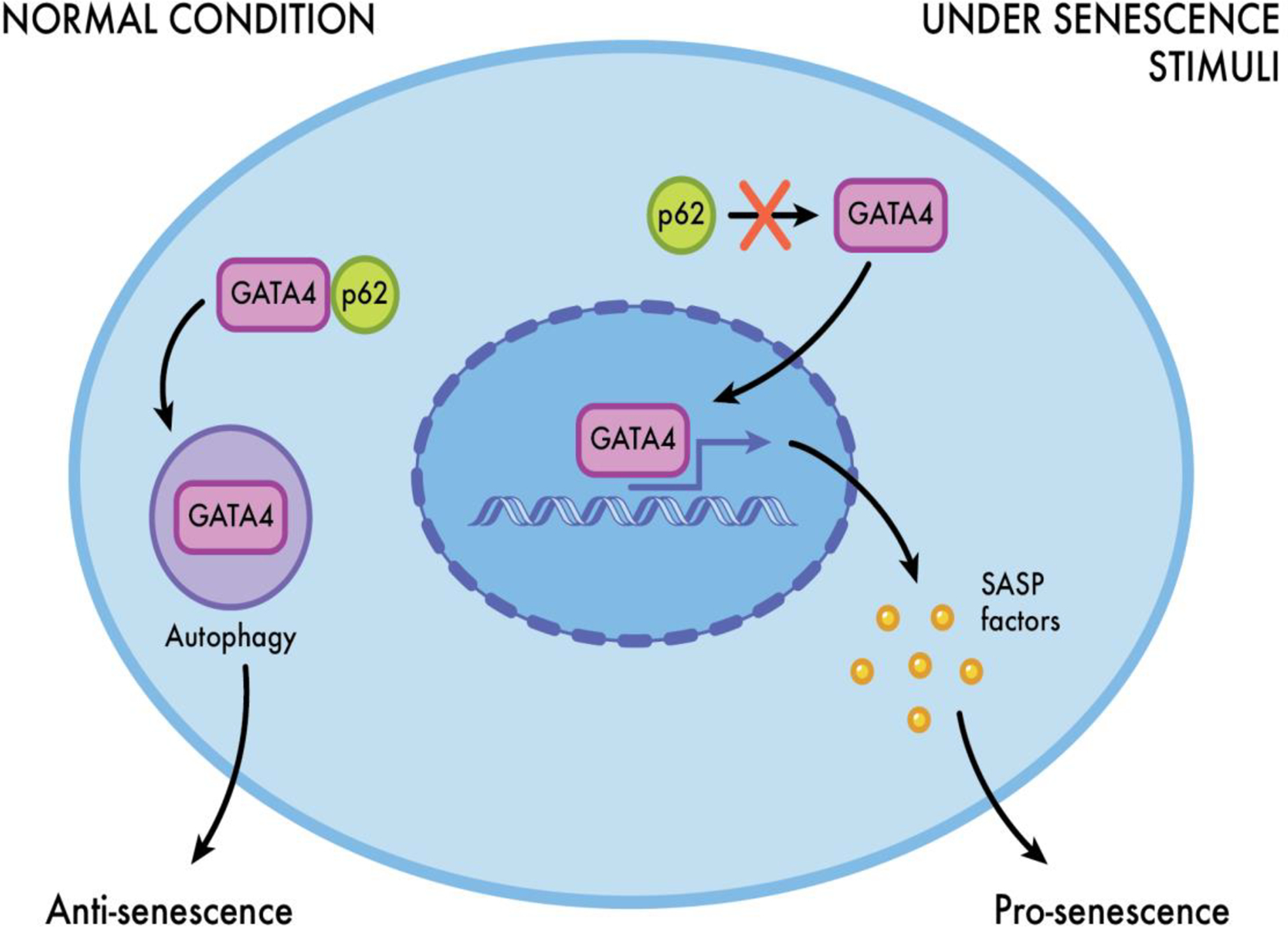
Depending on the cell type, selective autophagy can have antisenescence or prosenescence capabilities. In fibroblasts, selective autophagy tends to exhibit antisenescence capabilities, whereas, in primary lung cells, selective autophagy exerts prosenescence capabilities. However, in MSCs, selective autophagy presents antisenescence as well as prosenescence properties . For example, there is evidence suggesting that under normal conditions, p62 (autophagy receptor, sequestosome 1 [SQSTM 1]), binds strongly to GATA4, a central regulator of the SASP, which triggers GATA4 catabolism (selective autophagy) and prevents senescence in MSCs . In contrast, under senescence stimuli, p62 cannot bind to GATA4, and its selective degradation does not occur, resulting in the expression of cellular senescence ( Fig. 3 ). Ultimately, it seems that whether autophagy acts to induce or prevent cellular senescence depends on the type of autophagy, the timing, the stimuli triggering it and the cell type. Based on these complexities, additional research is needed to establish the role(s) of general and selective autophagy in MSCs and bone homeostasis and to potentially identify specific molecular targets for increasing health-span.
One caveat to many of the published studies addressing the role of autophagy in senescence is that the approach has been to typically target components of the autophagic core machinery such as Beclin (ATG6), LC3 (ATG8), or mTOR activity. As such, effects on general autophagy cannot be easily discerned from effects on selective autophagy pathways such as mitophagy. In order to fully elucidate whether autophagy is a prosenescence or antisenescence process will require a determination of the specific autophagy pathway and its functional role in the cells being studied. For example, during mitophagy, the core autophagic machinery is needed to catabolize damaged mitochondria and reduce ROS generation. Therefore, targeting any of the genes required for the core machinery would affect both general and selective autophagy. Thus, it would be necessary to target proteins that are dedicated to mitophagy, such as PTEN-induced putative kinase PINK1 and Parkin, an E3 ligase. The PINK1/Parkin axis is involved in targeting mitochondria with reduced membrane potential with polyubiquitin chains required for mitochondria targeting to autophagic vesicles ( Fig. 3 ). Once ubiquitinated, specific receptor proteins, including optineurin, NDP52, and TaxBP1, will recruit autophagic membranes to engulf these tagged mitochondria. The importance of PINK1 and Parkin in regulating mitochondrial function via mitophagy to prevent senescence also has been demonstrated in familial-age related diseases like Parkinson’s . However, not all mitophagy is dependent on PINK1/Parkin function. A recent study conducted in skeletal muscle cells identified AMPK as the key regulator of mitophagy by enhancing mitochondrial fission (via MFF phosphorylation) and mitochondrial autophagosome engulfment (via TBK1 phosphorylation) in a PINK1/Parkin independent manner . A role for AMPK in the regulation of mitophagy was previously indicated , and involves its role in inducing mitochondrial fission, a required event that precedes mitophagy, and in activating ULK1 which recruits damaged mitochondria to the lysosome . In addition to regulation of mitophagy, AMPK has many cellular functions, including regulation of metabolism and cellular homeostasis, cell survival and death, and general autophagy. All of these AMPK functions have the potential to impact aging. Therefore, it is not surprising that AMPK activation has been shown to delay aging and increase life span in Drosophila melanogaster and protect against senescence in aged rats by induction of autophagy that ultimately reduced oxidative damage .
AMPK and cellular senescence of MSCs
Multiple studies have indicated that AMPK activation prevents senescence in various cell types and that the mechanism involves AMPK-mediated activation of autophagy. In fibroblasts, AMPK activators like metformin prevented the development of oxidative stress-induced senescence. The prevention of senescence in the fibroblasts was due in part to increased autophagic flux NAD + homeostasis . Mechanistically, the results of the study by Han et al. are entirely consistent with the roles of AMPK in the regulation of cellular metabolism, general autophagy and mitochondrial homeostasis by mitophagy . AMPK is essentially a cellular energy sensor. It is a heterotrimeric complex composed of a catalytic α subunit and regulatory β and γ (gamma) subunits with multiple isoforms of each subunit encoded by different genes. The gamma subunit is responsible for the ability of AMPK to respond to changes in the ATP-to-AMP ratio via its ability to bind adenine nucleotides. The binding of AMP or ADP stimulates AMPK, to the gamma subunit via multiple mechanisms that have been recently reviewed . Once bound by AMP, activated AMPK will typically stimulate catabolic pathways such as glycolysis and fatty acid oxidation and autophagy, and block anabolic pathways such that ATP production is stimulated . Concerning general autophagy there may be multiple mechanisms whereby active AMPK can initiate autophagy or regulate autophagic flux. One of these mechanisms involves AMPK-mediated phosphorylation and activation of ULK1, a major effector of autophagy initiation. Of interest, the PGC-1α/AMPK pathway appears to play a critical role in preventing cellular senescence in MSCs.
PGC-1α is a master regulator of mitochondrial biogenesis that can be finely tuned in response to cellular metabolic fluctuations. AMPK α is one of the metabolic sensors that affect PGC1α activity and ultimately mitochondrial activity, defining a critical regulatory network for metabolic homeostasis . Further studies have identified C1q and tumor necrosis factor-related protein 9 (CTRP9) as a novel cytoprotective cytokine with antioxidant effects that may play a critical role in preventing cellular senescence and facilitating stem cell rejuvenation. Specifically, CTRP9 ameliorates cellular senescence via PGC-1α/AMPK signaling in the MSCs .
Overall it is clear that AMPK can play a central role in retarding senescence, most likely via its participation in cellular autophagy, and mitophagy pathways. Although activating AMPK with drugs such as metformin may be an attractive approach toward postponing senescence in cells of tissues, some caution is advised. For example, AMPK activation has been shown to support the viability of dormant breast cancer cells .
Antiaging effects of senostatics (senomorphics) in the musculoskeletal system
Not long ago, senescent cells were viewed as merely dormant cells which posed no noxious effects to human health. However, recent studies have demonstrated that senescent cells cause detrimental effects like systemic inflammation due to their SASP proinflammatory and prosenescent products. These have been shown to induce aging-related changes and are associated with many chronic diseases that come with age itself, such as osteoporosis and sarcopenia. Nonetheless, senescent cells also provide beneficial actions like protecting against cancer and assisting in wound healing . Indeed, it would not be wise to eliminate every senescent cell because they do serve a beneficial role in maintaining homeostasis. Rather a less drastic approach might be preferable and more helpful to increase the human health-span . Recently, agents have been identified with the capacity to inhibit proinflammatory products produced by the SASP. These include senostatics or senomorphics, which target processes in senescent cells which help drive the SASP or target subsets of SASP molecules directly. Among these targets are the rapamycin receptor (mTORC1) and the JAK1/JAK2-STAT3 pathway.
A few senostatics are already being studied with mouse models, including rapamycin inhibitors, Ruxolitinib (JAK1/JAK2-STAT3 inhibitor), glucocorticoids and metformin (commonly used to treat type 2 diabetes) . These agents have been shown to reduce frailty, heart failure and cognitive impairment in mouse models, suggesting the possibility of being able to increase health-span . Ruxolitinib, for example, is a JAK1/JAK2-STAT3 inhibitor. It is used clinically to treat polycythemia vera, myelofibrosis and graft versus host disease . In recent studies done with 22 and 24 months old C57BL/6 mice, it was found that Ruxolitinib has senostatic properties capable of attenuating insulin resistance, reduce osteoporosis, and decreased stem cell dysfunction , all effects with major significance in osteosarcopenia. Another study confirmed these results when mice treated with Ruxolitinib presented higher bone mass, better bone microstructure and improved cardiovascular function, suggesting the senotherapeutic agent may even be beneficial in multiple age-related comorbidities . Notably, the drug has proven to reduce frailty in human patients, confirming that the senomorphic properties of the drug are not only seen in mouse models .
The effectiveness of Ruxolitinib as a senostatic agent lies in its ability to inhibit the JAK/STAT pathway. This pathway can be overexpressed in senescent cells, and its inhibition leads to the suppression of the SASP proinflammatory products, ultimately reducing chronic systemic inflammation . It appears that the SASP between different cell types tends to be relatively similar . This is advantageous because it facilitates the development of new senotherapeutic agents that can simultaneously target SASP in multiple cell types. More importantly, it is possible that future senostatics can effectively reduce polypharmacy, one of modern medicine’s most significant drawbacks, by targeting and treating multiple chronic diseases concurrently.
Senolytics antiaging effects in bone
In 2015, additional senotherapeutic agents were discovered that killed senescent cells instead of inhibiting the SASP proinflammatory products . These agents were termed senolytics and included small molecules, peptides, and antibodies that target the senescence-associated antiapoptotic pathways (SCAPs) . Several SCAPs include the dependence receptors/tyrosine kinases, the BCL-2 family, the PI3K/AKT metabolic network, p53/p21/serpins, and HIF1 α . Interestingly, HIF1α competes against the aryl hydrocarbon receptor that is activated by kynurenine in MSCs, inducing senescence . These SCAPs inhibit the proapoptotic pathways of senescent cells, hence, blocking apoptotic cell death . Apoptosis, defined as programmed cell death, is a homeostatic mechanism that cells employ to regulate cell populations in tissues . Interestingly, researchers found that SCAPs can involve protein families, which are essential for maintaining the antiapoptotic pathways like tyrosine kinases (or dependence receptors) and the BCL-2 protein family . Thus, SCAPs reflect the diversity of composition of SASP that can depend on the senescent cell type, the stimulus triggering senescence, and the hormonal and growth factor milieu. Although SCAPs are cell and context dependent, the targeting (blocking) of the functioning SCAPs permit the proapoptotic pathways to be expressed and ultimately induce apoptosis of the senescent cells .
The first effective senolytic drug recipe (D + Q) combines Dasatinib, an FDA approved antibody-based tyrosine kinase inhibitor, and Quercetin, a flavanol present in many fruits and vegetables . Independently, Dasatinib works by inhibiting the dependence receptors/tyrosine kinase SCAP and Quercetin by targeting the BCL-2/BCL-XL, PI3K/AKT, and p53/p21/serpine SCAPs. Initial studies showed that Dasatinib was able to block the SCAP of primary human senescent preadipocytes but not the SCAP of primary human embryonic venous endothelial cells HUVECs. In contrast, Quercetin effectively blocked the SCAP in the senescent HUVECs, but not the SCAP of the senescent preadipocytes. Importantly, D + Q used in combination eliminated both the senescent preadipocytes and HUVECs. A similar synergistic senolytic outcome was noted when D + Q was used to treat cultured senescent mouse embryonic fibroblasts, compared to the complete lack of senolytic activity when D + Q were used separately .
Research on human adipose tissue explants found that the senescent cells could be eliminated after 18 h of continuous exposure to D + Q treatment . Even more astonishing is the fact that albeit D + Q has a half-life of only a few hours, it was able to diminish the detrimental effects of exposing a mouse leg to a 10-Gy dose of radiation, even 7 months after injury . In more recent studies, D + Q treatment has been shown to alleviate osteoporosis in mice; however, this outcome required intermittent dosing of the D + Q combination . This may occur because intermittent senolytic treatment effectively allowed osteoprogenitors cells like osteoblasts to maintain function. It is known that SASP produced by senescent cells disrupts the balance between bone deposition and resorption, with more bone degradation and, ultimately, calcium in the bloodstream. Over time, this disruption in bone homeostasis can lead to degenerative bone diseases like osteoporosis. Thus, by eliminating the senescent osteocytes with senolytic drugs, the SASP molecules are also eradicated. Consequently, the quantity of osteoblasts increases and bone formation follows shortly after . The fact that intermittent dosing was required to effectively treat osteoporosis in this murine model underscores the fact that the exact dosing of a senolytic drug will depend at least in part on the rate of senescent cell accumulation .
Recently, two additional senolytic agents were discovered. One is a powerful flavonoid called Fisetin, while the other is an antitumor agent named navitoclax, which is a BCL-2 family inhibitor. Bcl2 family molecules are part of the larger binding homology domain (BH3) family because each member of this family contains a BH3 domain critical for regulating apoptosis. Navitoclax is a BH3 mimetic that potently binds to the BH3 domain of Bcl2 and BCLXL , and effectively inhibits the BCL-2/BCLXL SCAP allowing the proapoptotic pathways to kill the senescent cells expressing these BH3 family members .
A major setback in the development of new senolytic drugs is the fact that SCAPs tend to differ between cell types . This makes individual senolytic drugs somewhat specific and only effective toward subsets of senescent cells. An example of this is navitoclax, which is effective against senescent HUVECs, but ineffective against primary human adipocyte progenitors . However, in advanced states of tissue senescence, the effectiveness of senolytics may be an issue. For example, we have recently shown that with advanced age in mice, the degree of senescence in bone may be so high that treatment with a senolytic like navitoclax may actually increase bone loss due to killing too many osteogenic cells leading to further inhibition of bone formation/repair . Therefore, in clinical practice, it may be problematic to use senolytics or careful dose titration and targeting earlier phases of the disease process with a lower degree of overall senescence may be needed. In addition, whether these therapies also work in sarcopenia remains unknown.
The field of senolytics has drawn much attention from the scientific community, given the potential antiaging properties of these senotherapeutic drugs. This interest only grew more intense following research with the INK-ATTAC mouse model. These mice allow systemic knockout of the p16 Ink4a tumor suppressor gene after treatment with AP20187, a drug with senolytic properties . This pharmacological approach produced the elimination of both senescent and nonsenescent cells. In comparison, the other two experimental groups were treated with the senolytic D + Q and the senostatic Ruxolitinib . These latter two approaches also improved bone microarchitecture and strength, suppressed bone resorption, and promoted bone formation . Furthermore, the data collected from the study demonstrated an increase in osteoblast numbers and a decrease in bone marrow adipose tissue in old INK-ATTAC mice after treatment with AP20187 compared with vehicle-treated mice. These results are consistent with stem cells in bone tissue committing to the osteoblastic lineage instead of the adipocyte lineage, a phenomenon which tends to happen with aging and is thought to decrease the size of the osteogenic cell pool, consequently reducing the rate of bone formation leading to homeostatic imbalance and bone loss .
Senolytic drugs continue to be identified, and their use supports the concept that the elimination of senescent cells is a viable approach to treating chronic diseases of aging. An example is UBX0101, a senolytic agent that targets the BCL-2 family and can eliminate senescent cells in articular cartilage and synovium following anterior cruciate ligament transection (ACLT). This senolytic agent proved to alleviate osteoarthritis in mice . Another example is the combination of curcumin and 0-vanillin, which proved to reduce senescent intervertebral disk cells, reduce back pain and increase matrix synthesis . While there is ample evidence supporting the beneficial effects of senolytics, there is also evidence suggesting that many also pose cytotoxic effects, as is the case of the BCL-2 inhibitors . Moreover, many senolytic drugs lack specificity toward senescent cells and kill nonsenescent cells as well. Recently, however, a group of researchers identified a promising senolytic drug capable of selectively killing senescent cells. The drug, 17-DMAG, inhibits the heat shock protein (HSP90) . This chaperone protein is ubiquitously expressed in mammalian cells and assists in maintaining cellular senescence. It is critical for protein stabilization, including that of antiapoptotic proteins. For example, 17-DMAG disrupts the interaction of HSP90-AKT, hindering the antiapoptotic pathway PI3K/AKT . Without this essential survival pathway, many senescent cells are no longer viable and enter apoptosis. Importantly, 17-DMAG was found to have senolytic activity in-vivo as well as in-vitro. Furthermore, the compound has the capacity to selectively kill senescent cells induced to undergo senescence by different types of stimuli like DNA damage, genotoxic stress, oxidative stress, and inflammatory stress. Even more, the compound can broadly kill senescent cells from different sources, such as mouse fibroblasts, mesenchymal stem cells, and human lung fibroblasts. Of particular advantage, intermittent dosing was enough to reduce the symptoms of various age-related chronic diseases like neurodegeneration, frailty, ataxia, gait disorder, kyphosis, and osteoporosis. While there is still much to learn regarding senotherapeutics, senolytic drugs are potential candidates for treating bone degenerative diseases like osteoporosis, increasing health-span, and reducing polypharmacy.
Senostatics or senolytics
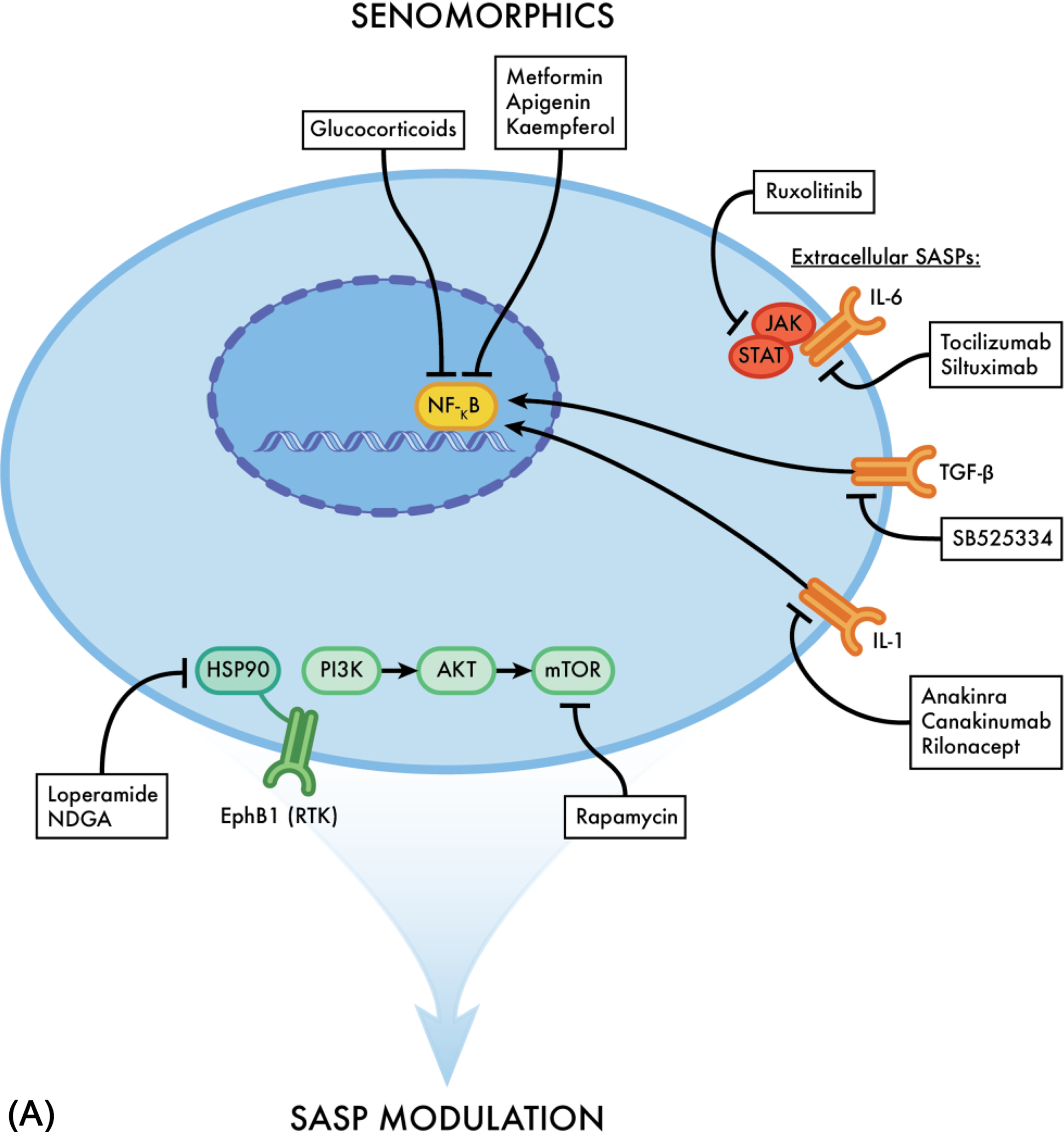
Senotherapeutic agents have drawn interest among the scientific community given their antiaging effects. Nonetheless, questions of whether senostatics (AKA senomorphics) should be used over senolytics, or in combination and the timing remain. In order to better understand if one has an advantage over the other, it is essential to understand the mechanisms of action for each and how they interact with senescent cells ( Fig. 4 A and B ). Evidence suggests that utilizing SASP inhibitors can alleviate many chronic diseases such as osteoporosis and sarcopenia, where age is a primary underlying risk, by inhibiting their proinflammatory products and reducing stem cell dysfunction ( Fig. 5 ). A few like Ruxolitinib (JAK/STAT pathway inhibitor) and metformin, most commonly used to treat Type II Diabetes have already shown favorable results in preclinical trials . Additional evidence suggests that utilizing senostatics over senolytics has the advantage of leaving the previously senescent cells intact, which sustain tissue structure and physiological integrity when a very high percentage of the tissue’s cells undergo senescence .
Related posts:
 Cellular senescence and other aging mechanisms in bone and muscle
Cellular senescence and other aging mechanisms in bone and muscle
 Biomechanics and mechanobiology of bone and muscle: Current and future musculoskeletal modeling research directions in osteosarcopenia
Biomechanics and mechanobiology of bone and muscle: Current and future musculoskeletal modeling research directions in osteosarcopenia
 Osteosarcopenic adiposity
Osteosarcopenic adiposity
 Sex steroids and gender differences in muscle, bone, and fat
Sex steroids and gender differences in muscle, bone, and fat
 Pharmacological management of osteosarcopenia
Pharmacological management of osteosarcopenia
 Frailty: The end of the osteosarcopenia continuum?
Frailty: The end of the osteosarcopenia continuum?
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


