Fig. 1
Mechanisms of MDR towards cancer chemotherapeutic drugs. Cancer cells can develop resistance to multiple drugs by various mechanisms as depicted. Mechanisms include (a) decreased uptake of drug, (b) reduced intracellular drug concentration by efflux pumps, (c) altered cell cycle checkpoints, (d) altered drug targets, (e) increased metabolism of drug, and (f) induced emergency response genes to impair apoptotic pathway [Reproduced from Chai et al. (2010) under the terms of Creative Common Attribution License]
A large number of chemical agents including calcium blocker, calmodulin inhibitors, coronary vasodilators, indole alkaloids, quinolines, hormones, cyclosporins, surfactants, and antibodies act as modulators of acquired MDR, which have the ability to reverse the drug efflux function of P-gp (Krishna and Mayer 2000). Toxicity is one of the significant issues reported in several clinical studies, as most of these modulators resulted in nonspecific toxic effects which are not considered acceptable during chemotherapy and prevents their safe use during treatment (Shukla et al. 2011). The use of natural agents in the realm of MDR holds potential as these display minimal toxicity to humans compared to conventional chemotherapies and have pleiotropic action mechanism that could target numerous signaling pathways. This is beneficial as malignant transformation and progression are multistage processes caused by gene alterations in more than one signaling pathway. Therefore, the impact of natural agents on cancer treatment could be more efficacious, as they can be used alone or as an adjuvant in standard chemotherapy to counter cancer cells’ defense mechanisms such as MDR (Abdallah et al. 2015). The present chapter provides a brief overview of the potential the natural compounds of dietary and herbal origin hold as promising candidates in overcoming drug resistance in cancer disease.
2 Mechanisms of MDR
A diverse range of molecular mechanisms have been implicated in drug resistance; these include reduced cellular uptake of drug, increased rates of drug efflux, alterations in drug metabolism (drug inactivation and elimination), altered expression of drug targets, epigenetic events, activation of survival signaling pathways, inhibition of downstream death signaling pathways, and the influence of the local tumor microenvironment (Fig. 2). Recently, the failure of chemotherapy in certain cases has been attributed to the presence of cancer stem cells, which are intrinsically highly resistant to many therapeutic approaches (Valent et al. 2012).
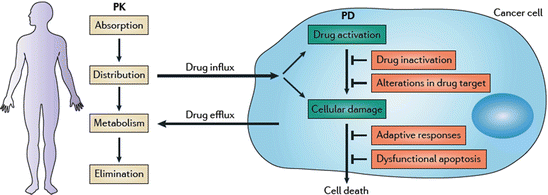
Fig. 2
General principles of drug resistance. Pharmacokinetic (PK) factors such as drug absorption, distribution, metabolism, and elimination (ADME) limit the amount of a systemically administered drug that reaches the tumor. In the tumor, the effects of the drug on the cancer cell are collectively termed its pharmacodynamic (PD) properties. The anticancer activity of a drug can be limited by poor drug influx or excessive efflux; drug inactivation or lack of activation; alterations such as changes in expression levels of the drug target; activation of adaptive prosurvival responses; and a lack of cell death induction due to dysfunctional apoptosis, which is a hallmark of cancer [Reproduced from Holohan et al. (2013) with permission of Macmillan Publishers Limited]
In case of certain solid tumors, angiogenesis is compromised (Jain 1987) leading to poor vasculatures that hinder the accessibility of the drug to the cancer cells thereby limiting the drug-induced cytotoxicity. The growth environment in which cancer cells proliferate is markedly different from that of the normal cells. Obstructed access of nutrition and hypoxia due to poor vasculature and the resultant lactic acid accumulation could confer resistance to cancer cells against drugs that act on actively dividing cells or the cellular uptake of which requires a pH gradient (Demant et al. 1990). However, the major factor observed to be responsible for multi drug resistance is the efflux of drugs across the plasma membrane by the ATP-binding (ABC) transporter family of transmembrane proteins. These are multidrug resistance protein-1 (MDR1) also known as P-glycoprotein and breast cancer resistance protein (BRCP). MDR1 over-expression has been associated with chemotherapy failure in many cancers, including kidney, colon, liver, prostate, lung, and breast cancers as well as in leukaemias and lymphomas (Holohan et al. 2013).
Drug inactivation is another mechanism of drug resistance induced by cellular xenobiotic metabolism, for example, inactivation of platinum drugs by thiol glutathione (Meijer et al. 1992). Epigenetic events that silence gene of key enzyme required for the activation of pro-drug such as methylation of gene encoding thymidine phosphorylase have also been attributed to drug resistance. An example of this kind is the resistance to capecitabine which is a prodrug that needs thymidine phosphorylase for conversion to active drug 5-FU (Kosuri et al. 2009). Alteration in drug targets such as increased expression of target proteins reduces the efficacy of inhibitors of these targets as more target molecule must be inhibited to have an effective outcome (Palmberg et al. 1997). Most of the anticancer chemotherapeutic drugs act to damage DNA in cancer cells in order to direct these cells to apoptosis. However, cancer cells have evolved mechanisms that alter the normal cell cycle and DNA repair machinery enhancing its repair capacity and also evade the phenomenon of apoptosis thereby rendering the drug ineffective (Bouwman and Jonkers 2012). The target-associated resistance has also been observed such as gatekeeper mutations in oncogenic kinases such as that of BCR-ABL1 and T315, associated with imatinib resistance in chronic myeloid leukemia (Weisberg et al. 2005). Furthermore, recent studies have shown that cancer stem cells which might be contributing to the majority of the incidence of relapse are armed with several of these critical features responsible for drug resistance and projects emerging challenge in the premises of multidrug resistance in cancer (Holohan et al. 2013). Thus, drug resistance may be regarded as a multifaceted and dynamic phenotype which ultimately results in enhanced tumor cell survival and reduced chemo-responsiveness, regardless of the specific mechanism(s) involved.
3 Pharmacological Modulators of MDR
The pharmaceutical agents that posses the ability to reverse the resistance against anticancer drugs are called MDR inhibitors, chemosensitizers, or MDR modulators (Kellen 2003). Most of these inhibitors are targeted against P-gp transporters which were considered to be the principal factor responsible for the multidrug resistance. Accordingly, they are classified as the first, second and third generation of MDR reversal agents (Ullah 2008), with the fourth generation modulators still in infancy.
First Generation MDR Agents
First-generation modulators include verapamil (calcium channel blocker), quinine (antimalarial), cyclosporine A (immunosuppressant), tamoxifen (anti-steroid), and erythromycin (Ford and Hait 1990). These drugs were not specifically developed for MDR inhibition but were used for other pharmacological interventions and coincidentally found to be effective in sensitizing the drug-resistant tumors towards chemotherapy. However, their low affinity for the transporter proteins required high doses to achieve the desired effect, which resulted in adverse effects due to enhanced toxicity to normal cells, thus undermining the overall impact on clinical management (Lampidis et al. 1986).
Second Generation MDR Agents
The second generation drugs included valspodar (a non-immunosuppressive analogue of cyclosporine A) and R verapamil (R-enantiomer of verapamil, a weaker calcium channel blocker) (Hollt et al. 1992), which were designed by modification of the first generation modulators. The modifications were aimed at reducing their adverse effects by eliminating their non-MDR pharmacological action, thereby making them specific for MDR. However, within few years, the need for better MDR drug candidates arise as second generation drugs also failed to deliver the desired range of efficacy due to their low affinity for their target transporter proteins.
Third Generation MDR Agents
The third generation inhibitors are designed specifically for high transport affinity and low pharmacokinetic interaction. These include tariquidar (anthranilamide derivative), biricodar (pipecolinate derivative), Annamycin (anthracycline derivative), mitotane (2,4-dichloro-diphenyldichloroethane derivative), zosuquidar (dibenzosuberane derivative), and laniquidar (benzazepine derivative) (Liscovitch and Lavie 2002). These compounds exhibit effective MDR modulatory potency, high affinity, and selectivity for target MDR transporter(s) at low nanomolar range and subsequently low toxicity towards normal cells.
It may be mentioned that first- and second-generation modulators compete as a substrate with the cytotoxic agent for transport by the P-gp pump. This limits the efflux of the cytotoxic agent, increasing its intracellular concentration. However, third-generation inhibitors of P-gp, such as tariquidar, are noncompetitive inhibitors that bind with high affinity to the pump but are not themselves substrates. This induces a conformational change in the protein, thereby preventing ATP hydrolysis and transport of the cytotoxic agent out of the cell, resulting in an increased intracellular concentration. Moreover, in response to cytotoxic agents, cytochrome P450 enzymes are also induced and aid in drug metabolism and clearance. Several of the second-generation P-gp modulators, including valspodar and biricodar, are substrates for this enzyme. These partially impair drug metabolism and elimination, significantly reduce the systemic clearance of anticancer drugs, and consequently elevate toxicity. The competition between cytotoxic drug and these P-gp modulators for cytochrome P450 3A4 activity has resulted in unpredictable pharmacokinetic interactions. MDR inhibitors such as valspodar block the cytochrome P450 3A4-mediated metabolism of paclitaxel and vinblastine resulting in increased serum concentrations of the cytotoxic agents and potentially placing patients at risk of cytotoxic drug overexposure (Thomas and Coley 2003). Third generation agents do not affect cytochrome P450 3A4 at relevant concentrations and therefore do not alter the plasma pharmacokinetics of the cytotoxic drug.
It is to be noted that an array of novel approaches are being conceived to circumvent MDR including inhibition of expression of ABC transporter by targeting mRNA through antisense oligonucleotides, hammerhead ribozymes, and siRNA in addition to transcriptional regulation, plasma membrane alteration, drug encapsulation, and antibodies (Wu et al. 2008). Novel strategy using improvised nanoparticle-based drug delivery system (DDS) has also been suggested to reverse the MDR in resistant phenotype (Fig. 3). As the cancer cells continuously evolve novel mechanisms of drug resistance which might impede the conventional therapy, a proactive approach is warranted to explore large number of MDR modulators with elevated margin of safety and desired range of efficacy. Many natural products of dietary and non-dietary origin have shown promising chemosensitizing effects on ABC drug transporters demonstrating broad-spectrum modulatory effects on more than one ABC drug transporter (Wu et al. 2008).
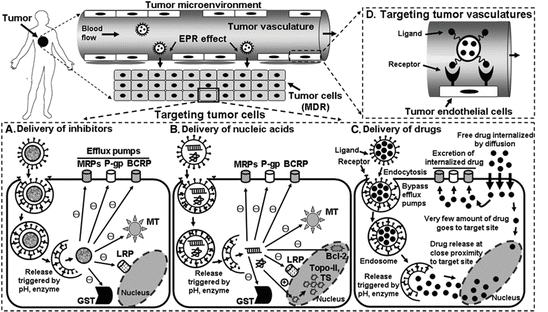
Fig. 3
Schematic representation of the application of a DDS for reversing cancer MDR. The expression of proteins or enzymes, responsible for MDR in cancer cells, can be controlled by delivering either a specific inhibitor or nucleic acids followed by the delivery of cytotoxic drug (either concurrently or separately) via a nanoparticle. The free drug, internalized by diffusion, can easily be detected by the ABC transporters and excreted out before going to the depth of the cells. Nanoparticles loaded with free drug can be endocytosed, thus permitting them to bypass the ABC transporters and deliver their payload to the target organelle where the drug exerts it action. These delivery approaches would reverse the MDR of cancer cells by making them chemosensitive [Reproduced from Kibria et al. (2014) with permission of Springer Science & Business Media]
4 Diet-Derived Factors as Potential MDR Modulators
Clinical experiences from the past have demonstrated that most of the agents from the first, second, or third generation of MDR modulators suffer clinically from their intrinsic toxicity or from undesired effects on the pharmacokinetics of the accompanying anticancer drugs (Holohan et al. 2013). These limitations have lead to continuous efforts to search for new and more productive compounds that could be effective at tolerable doses without any adverse effect. It is understood that on account of their routine intake and least toxicity, many of the natural products from fruits, vegetable, spices, and other dietary supplements are currently being investigated for their anticancer activities and their role as MDR modulators is thought to augment their efficacy against cancer (Ullah 2008). Furthermore, the enormous diversity of compounds derived from natural resources such as plants provide chemical scaffolds as lead compounds suitable for development of novel inhibitors.
The earliest observation that elicited great interest in studies related to dietary modulation of drug transporters originated from study that reported the impact of active components of fruit extracts on the outcome of clinical treatment using P-gp drug substrates (Bailey et al. 1991). Since then a large number of dietary and non-dietary plant products have been the subject of MDR studies. Flavonoids, which are the widely distributed natural constituents of human diet, have been reported as drug transporters inhibitors (Yarla and Ganapaty 2013). Apigenin, biochanin, chrysin, daidzein, epigallocatechin (EGC), epigallocatechin-3-gallate (EGCG), fisetin, genistein, hesperetin, kaempferol, luteolin, morin, myricetin, naringenin, naringin, phloretin, phloridzin, quercetin, silibin, and silymarin are the different kinds of flavonoids which have been reported as BCRP inhibitors. Among these Chrysin and biochanin A are the most potent BCRP inhibitors, producing significant increases in mitoxantrone accumulation in BCRP over-expressing cancer cell lines (Zhang et al. 2004). The diverse structure of flavonoids has been subjected to various chemical modifications in order to obtain better P-glycoprotein inhibitors. In general, it was found that modifications that increased hydrophobicity of the molecule such as prenylation or geranylation significantly increased the modulatory activity of flavonoids (Kitagawa et al. 2005).
Related posts:
 Sense of Antioxidant–Pro-oxidant Conundrum in Anticancer Research
Role of Organosulfur Compounds Derived From Allium Vegetables in Cancer Prevention and Therapy
Role of Dietary Polyphenols in Prostate Cancer Chemoprevention
for Sesame Seed-Derived Factors to Prevent Colorectal Cancer
Sense of Antioxidant–Pro-oxidant Conundrum in Anticancer Research
Role of Organosulfur Compounds Derived From Allium Vegetables in Cancer Prevention and Therapy
Role of Dietary Polyphenols in Prostate Cancer Chemoprevention
for Sesame Seed-Derived Factors to Prevent Colorectal Cancer
 Role of Chemokine Receptor Signaling Axis and Natural Bioactive Chemopreventive Agents in Metastasis of Breast Cancer
Role of Chemokine Receptor Signaling Axis and Natural Bioactive Chemopreventive Agents in Metastasis of Breast Cancer
 in the Development of Black Seed-Derived Anticancer Agents
in the Development of Black Seed-Derived Anticancer Agents
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


