Fig. 6.1
a Multi-target sDNA sensitivity by colorectal cancer (CRC) stage. Sensitivity for screenrelevant CRC (stages I—III) was 97 % overall (74 of 76), 100 % for lesions at or proximal to the splenic flexure (proximal), and 94 % for lesions distal to the splenic flexure. b Multi-target sDNA sensitivity for advanced precursor lesions (advanced adenoma + sessile serrated adenoma) increases with increasing lesion size. This parallels the occurrence of high-grade dysplasia (HGD); 94 % of HGD (16 of 17) occurred in adenomas > 2 cm, and sDNA sensitivity for HGD was 83 % (15 of 18). sDNA stool DNA. (Adapted from Ref. [28]. With permission from Elsevier)
Table 6.1
Next-generation stool DNA test performance in case-control studies. (Reprinted from Ref. [56]. With permission from Sage Publications)
Sensitivity (%) | Specificity (%) | ||
|---|---|---|---|
CRC | Adenoma > 1 cm | ||
Prototypes | |||
Ahlquist et al. [24] | 85 | 64 | 90 |
Ahlquist et al. [25] | 91 | (82a) | 91 |
Optimized assay | |||
Lidgard et al. [26] | 98 | 64 | 91 |
Lidgard et al. [27] | 98 | 60 | 90 |
Validation in the Screen Setting: Pivotal Multicenter Cross-Sectional Study
A pre-commercial version of the optimized and automated MT-sDNA test (Cologuard; Exact Sciences, Madison, WI) has now been validated in a multicenter study on > 10,000 patients from the screen setting [30]. The study was based on a cross-sectional design in which the MT-sDNA test was compared to a commercial FIT using colonoscopy as the criterion standard (ClinicalTrials.gov Identifier: NCT01397747). The Cologuard MT-sDNA utilizes a quantitative allele-specific real-time target and signal amplification (QuARTS) assay of mutant KRAS, methylated BMP3, methylated NDRG4, and unmethylated β-actin as well as an immunochemical test for hemoglobin (Fig. 6.2). Cutoff values were preestablished using an analytic regression algorithm to determine an overall test score [22].
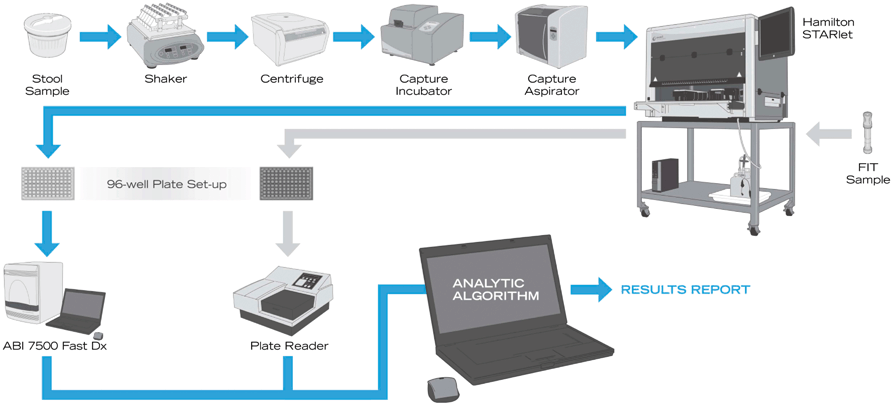
Fig. 6.2
The exact sciences automated analytic platform. Multi-target sDNA analytic process. Patient samples are homogenized in the collection container, aliquoted, and centrifuged. DNA markers are captured with target-specific magnetic beads (capture incubator), washed (capture aspirator), and magnetically separated. Bead-bound sDNA biomarkers are transferred to a Hamilton Microlab STARlet (Reno, NV). The portion of sDNA for methylation assay is bisulfite treated, and the portion for KRAS mutation assay is pH neutralized. Treated DNA is then combined with reagents for quantitative allele-specific realtime target and signal amplification (QuARTS) on an ABI 7500 FastDx (Carlsbad, CA) that generates results in log strands of DNA. Fecal hemoglobin samples are transferred to enzyme-linked immunosorbent assay plates with Hamilton Microlab STARlet liquid handler and then read (BioTek ELx808 plate reader, Winooski, VT). Software algorithmically integrates results of assays to calculate a dichotomous “positive” or “negative” result. (Adapted from Ref. [28]. With permission from Elsevier)
At predetermined cutoffs, Cologuard detected significantly more lesions than did FIT for all categories: 93 % versus 73 % for curable-stage CRC (Fig. 6.3a); 68 % versus 45 % for adenomas with high-grade dysplasia (Fig. 6.3b); 66 % versus 42 % for adenomas ≥ 2 cm, 42 % versus 24 % for all advanced adenomas, 17 % versus 8 % for non-advanced polyps (Fig. 6.3d); and, 43 % versus 5 % for sessile serrated polyps ≥ 1 cm (Fig. 6.3b). As we have previously noted [21], CRC detection was not affected by stage or site (Fig. 6.3c), and polyp detection increased in proportion to lesion size (Fig. 6.3d). Based on patients with normal colonoscopy, specificity was lower with Cologuard at 90 % compared to FIT at 95 %. Age influenced Cologuard specificity which was 94 % in those < 65 years of age and 87 % in those ≥ 65 years of age. Given the known variability in colonoscopy detection rates, it is possible that some of the “false-positive” stool test results represent lesions missed by the criterion standard. In addition to these pivotal trial data, rigorous technical studies demonstrate reliability of Cologuard performance in compliance with the highest regulatory standards for diagnostic testing.
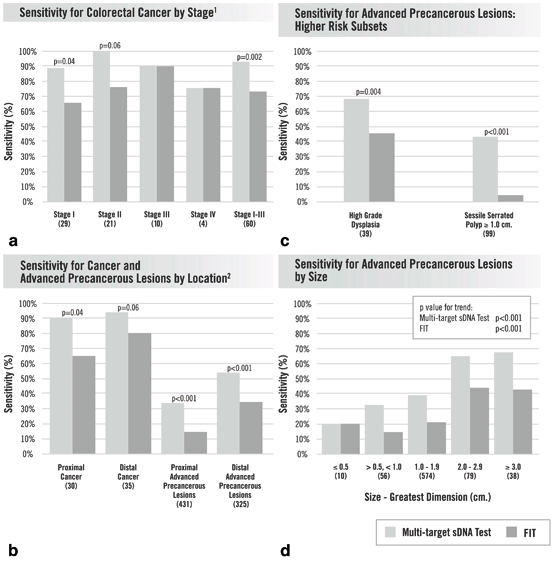
Fig. 6.3
Results of the multi-target sDNA test and commercial fecal immunochemical test in analyzed subgroups. Numbers in parentheses ( ) are sample sizes. a sensitivity for colorectal cancer by stage (1 The stage of one colorectal cancer was not available), b sensitivity for cancer and advanced precancerous lesions by location (2 The location of one advanced precancerous lesion was not available), c sensitivity for advanced precancerous lesions: higher risk subsets, d sensitivity for advanced precancerous lesions by size. (Adapted from Ref. 30. With permission from Massachusetts Medical Society)
Technical Quality and Regulatory Pathways
To better understand the quality benchmarks required for CRC screening by sDNA, the regulatory mechanisms of new in vitro diagnostic tests deserve further review. Broadly speaking, there are two major pathways for new tools to enter the laboratory medicine marketplace in the USA; these are Clinical Laboratory Improvement and Amendment (CLIA) certification and/or premarket approval (PMA) by the Food and Drug Administration (FDA) [31]. Tests developed by a single laboratory for use at a single site are historically regulated by the Centers for Medicare and Medicaid Services (CMS) under the CLIA Act of 1988 [31]. The focus of CLIA certification is analytical test accuracy. While quality control metrics are reviewed, the requirements for demonstration of clinical effectiveness and clinical validation are less strictly defined by this approval mechanism. In contrast, tests intended for broader licensure or distribution are subjected to a more rigorous premarket demonstration of safety and efficacy by the FDA . Within the FDA, a diagnostic device that bears substantial equivalence to another cleared predicate product may qualify for premarket notification (e.g., 510 (k) review). In other cases, where no predicate exists or the test is a new device, the US Code of Federal Regulation (CFR) requires PMA. The extensive and detailed evidence needed to support a PMA is defined in Code of Federal Regulations 21 CFR, part 814.
The FDA encourages early engagement and collaboration in the PMA process [32]. For example, clinical trials are usually needed to generate the evidence required to establish safety and efficacy. Prior to clinical studies of unapproved diagnostics or other medical devices, manufacturers may interact with the FDA to establish trial objectives and endpoints required to substantiate approvability. This interaction is referred to as the pre-Investigation Device Exemption (IDE) process [31]. In the pre-IDE, the FDA may help prioritize scientific issues required for the PMA and perform preliminary review of the device technology. In the pre-IDE process for Cologuard, the FDA determined that FIT was not a clear predicate test because DNA is a fundamentally different analyte than hemoglobin, and the overall MT-sDNA assay process is substantially more complex. In addition, Cologuard required demonstration of safety and effectiveness in the average-risk screen setting, subject to accepted standards for reporting diagnostic accuracy [33]. Therefore, a PMA was required.
In addition to demonstration of safety and efficacy, a PMA requires submission of extensive technical or “nonclinical” data according to FDA guidelines, including careful description of the diagnostic test device, quality controls on all analytical reagents and assay equipment, laboratory analytical performance, prespecified cutoffs, and the technical limits of assay detection. The automated assay equipment for high-throughput testing (Fig. 6.2) [28] was subjected to similar rigorous verification and validation.
The optimized Cologuard MT-sDNA assay [22] was designed, defined, and reviewed a priori to any final screen setting clinical validation. The test then was subjected to tests of reproducibility in three independent clinical labs, each using the final, fully automated assay platform. At each lab, two independently operating teams performed the assay over 20 work shifts.
Nonclinical testing also studied the potential for artifactual assay interference or inhibition. Samples were pooled to achieve specific Cologuard cutoff scores and included both positive and negative cases as well as controls. Interference was tested separately for both the DNA and hemoglobin-based components of the assay. Prior to assay, samples were spiked with up to 56 items in nine classes from commonly ingested foods and medications including sources of animal hemoglobin and DNA, dietary fiber supplements, nonsteroidal anti-inflammatory drugs, and numerous other drugs.
Because methylation of BMP3 and NDRG4 has been reported in association with other disease processes, [34, 35] studies were performed to rigorously measure assay specificity and cross-reactivity. Samples from patients with inflammatory bowel disease (IBD) and other gastrointestinal and genitourinary cancers were tested. While some cross-reactivity with liver and pancreatic cancers was anticipated [36], these would account for much fewer than 1 % of false positives.
Multivariate analysis of demographic and anthropomorphic variables and patient exposure characteristics demonstrated that there was measurable influence from age on methylated DNA marker levels [37]. However, sex, race, body mass index, alcohol use, and smoking history did not impact test results. Importantly, family history of CRC and personal history of colorectal neoplasia had no significant influence [37].
The FDA also had input on methods to provide the appropriate clinical evidence for Cologuard, and suggested design elements for a prospective clinical study of average-risk patients in the screen setting was required (see DeeP-C study results, above) . The PMA submission to the FDA , including results of DeeP-C, was subsequently completed in the summer of 2013. On the basis of the clinical performance and safety demonstrated in the DeeP-C study, the FDA advisory panel unanimously endorsed Cologuard on March 27, 2014. On August 11, 2014, Cologuard became the first device to obtain full FDA approval and a simultaneous proposed national coverage determination from CMS.
Program Performance and Effectiveness
Based on what is known about the natural history of colorectal neoplasm progression and the point-in-time performance of the Cologuard test, models can be developed to derive an optimal testing frequency. While not finalized yet, our early modeling suggests that an interval of every 3 years could reduce CRC mortality and incidence at rates comparable to those by colonoscopy done at an interval of every 10 years. Based on early-generation sDNA data, the American College of Gastroenterology has also agreed that sDNA testing be implemented at 3-year testing intervals [38]. Furthermore, CMS coverage was also approved for this interval.
Program Sensitivity
CRC prevention (i.e., incidence reduction) depends on how effectively over time a screening program identifies and removes those premalignant lesions at greatest risk for cancer progression. Observational studies suggest that the majority of 1 cm polyps do not progress and polyps that do progress grow slowly with average doubling times of every 4–6 years [39–41]. Further, we noted in a recent study that > 90 % of all high-grade dysplasia was found in adenomas ≥ 2 cm, a size range where MT-sDNA sensitivity is high [28]. As such, longitudinal screening by Cologuard may yield excellent cumulative sensitivity for a cohort of advanced adenomas as they slowly grow and progress through high-grade dysplasia (Table 6.2). In contrast, CRC mortality reduction mandates very high point sensitivities for early-stage CRC; one cannot rely on repeated screening for effective detection as the window of cancer progression from early-to-late stages may be narrow [42]. With validated sensitivity for curable-stage CRC of 93 % in the screen setting, Cologuard noninvasively achieves detection rates similar to those of colonoscopy and meets this requisite of high curable-stage CRC detection (Table 6.2).
Table 6.2
Hypothetical programmatic sensitivity of MT-sDNA testing at a 3-year screening frequency
Cumulative sensitivity (%) | ||
|---|---|---|
Adenoma > 1 cma | Curable-stage CRC | |
Screen 1 | 42–66 | 93 |
Screen 2 | 71–91 | 99 |
Screen 3 | 90–98 | – |
Program Specificity
Nonspecificity in CRC screening tests leads to false-positive results and, consequently, to unnecessary and costly colonoscopies. From a program standpoint, it is cumulative false positives over time (or the rate of false positives) that is most relevant. The false-positive rate is a function of both specificity and testing frequency. Cologuard, with a point specificity of 87–90 % (10–13 % false-positives), applied at a frequency of every 3 years would yield an average rate of 3.3–4.3 false positives per year. FIT with a point specificity of 95 % performed at an annual frequency would yield 5 % false positives per year. Thus, the program specificity of an MT-sDNA test could prove to be comparable to or even higher than that of FIT.
Perceived Value in an Accountable Care Era
Perception of value is an important driver of test choice. Yet, a screening test’s value may be differently perceived across the various stakeholders, including patients, health-care providers, third-party payers, employers, industry, or society as a whole. For example, a reimbursement perspective of a CRC-screening approach may not account for patient inconvenience and costs (such as disrupted daily routines, missed work, and time and expense of travel) or for societal costs (such as lost work productivity required for screening). A reimbursement perspective may differ dramatically between cradle-to-grave lifetime coverage programs and those that cover only specific patients, such as Medicare beneficiaries. As such, cost-effectiveness and other value models may reach substantially different and competing conclusions based on the same available data [43].
Let us first consider the perspective of reimbursement for Medicare beneficiaries. The goal of the CMS review was to determine whether or not a new innovation is reasonable and necessary for the Medicare population to justify a national coverage decision (NCD) [44]. While the CMS review process utilized FDA assessments of safety and efficacy, FDA approval alone was not sufficient for the NCD. In this process, CMS requires an explanation of the relevance of the evidence presented for the NCD and the rationale for how this evidence demonstrates medical benefit specific to the Medicare population. The DeeP-C trial was designed to address these questions; of the patients enrolled in DeeP-C, 61 % were over the age of 65, and therefore MT-sDNA performance can be specifically assessed in those potentially eligible for Medicare coverage.
Related posts:
 Toward a Learning Health-Care System: Use of Colorectal Cancer Quality Measures for Physician Evaluation
Managing Quality in Ancillary Services
Medical Legal Aspects of Quality Improvement
Areas for Future Research
Toward a Learning Health-Care System: Use of Colorectal Cancer Quality Measures for Physician Evaluation
Managing Quality in Ancillary Services
Medical Legal Aspects of Quality Improvement
Areas for Future Research
 Quality Indicators and Benchmarks for Guideline-Recommended Fecal Occult Blood Tests
Quality Indicators and Benchmarks for Guideline-Recommended Fecal Occult Blood Tests
 Quality Indicators for CT Colonography
Quality Indicators for CT Colonography
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


