Principles of Breast Cancer Genomics
 ABSTRACT
ABSTRACT
Breast cancer is a disease characterized by clinical and molecular heterogeneity. Microarray-based gene expression profiling has identified distinct gene expression profiles associated with specific clinicopathologic features as well as differing prognosis and response to therapy. Several multiparameter assays are commercially available, and their clinical utility is being further evaluated in prospective clinical trials. Newer technologies now permit evaluation of not only gene expression but also single nucleotide polymorphisms, gene copy number, genomic mutations, and epigenomic or microRNA patterns that modulate gene expression. These state-of-the-art molecular approaches offer new insights into the pathogenesis of breast cancers and offer the promise to improve the prevention, diagnosis, and management of the disease.
Keywords: breast cancer, genetic, epigenetic, gene expression profiling, microarray technology, genomic instability, sequencing, methylation, comparative genomic hybridization, histone acetylation, MIAME, mRNA, miRNA, non-coding RNAs, RNA splicing, microRNA processing, FFPE, the cancer genome atlas, genomic assays
 INTRODUCTION
INTRODUCTION
Genomics is defined as the study of all of the nucleotide sequences in an organism. Cancer is characterized by cumulative genomic and epigenomic alterations induced by both endogenous and exogenous factors (1). For the most part, “genomic profiling” has focused on the evaluation of gene expression or the translation of the information encoded in genomic DNA into an RNA transcript. RNA transcripts include messenger RNAs (mRNAs) which are translated into proteins and various other RNAs (e.g., transfer RNA, ribosomal RNA, microRNA [miRNA], and noncoding RNA) that have important biologic functions. Although molecular profiling in breast cancer has focused largely on gene expression, the same principles may be applied to the study of the epigenome (2, 3), miRNA (4), proteins (5), or integrative approaches that evaluate combinations of profilingmethods (6). This requires bioinformatics which permit analysis and interpretation of the large amount of data generated (7). By combining high-throughput specimen evaluation and sophisticated bioinformatics analysis, one can identify distinctive patterns of expression which correlate with clinical behavior or response to specific therapies. Some have referred to these distinctive expression patterns as molecular “portraits” (8) or “signatures”(9), and the assay used to detect these patterns as “multiparameter assays”; the latter term has been used because rather than relying on expression of a single gene or protein, these assays typically incorporate information from measuring expression of multiple genes by using mathematical algorithms to derive a qualitative (e.g., high vs low risk) or quantitative (e.g., score) test result (10). The promises and pitfalls in developing multiparameter assays have been reviewed elsewhere (11–14), and specific criteria have been proposed for the level of evidence required to define and support their clinical utility (15). Several multiparameter assays are currently approved for clinical use, including some which have been recommended by expert panels for clinical decision making (10, 16). In this review, we will provide a broad overview of methods used to characterize the genomics of cancer and application of those methods to breast cancer, including a glossary of common terms that are summarized and described in Table 1.
Terms commonly used in describing microarray studies
Term | Definition |
General terms | |
Genomics | Study of all of the nucleotide sequences, including structural genes, regulatory sequences, and noncoding DNA segments in the chromosomes of an organism |
Genomic instability | Alterations in genomic DNA that are almost always associated with cancer |
Microsatellites | Small repetitive DNA regions, usually spanning 1–5 base pairs and repeated 15–30 times |
Microsatellite instability | Altered microsatellite sequences throughout a cell genome which contribute to nucleotide sequence alteration and allelic loss |
Chromosomal instability | Alteration in chromosomal number |
Loss of heterozygosity | Loss of function of one allele of a gene when the other allele has been inactivated |
Single nucleotide polymorphisms | DNA sequence variation occurring in a single nucleotide (A, C, T, or G) |
| Mechanisms of DNA repair | |
Homologous recombinatio | Repair of double-stranded DNA breaks by exchanging nucleotide sequences between two similar or identical strands of DNA |
Nonhomologous end joining | Repair of double-stranded DNA breaks by directly ligating without the need for a homologous template |
Base excision repair | Removal of small, non-helix-distorting base lesions |
Nucleotide excision repair | Repair of chemical and UV-induced DNA damage |
Translesion synthesis | Permits DNA replication past DNA lesions (e.g., thymidine dimers, AP sites) |
| Methods for evaluation of genome | |
Comparative genomic hybridization | Method used to evaluate variation in DNA copy number, including chromosomal loss, deletion, gain, or amplification |
DNA microarray | Method for evaluating gene expression which uses a glass slide or silicon chip with DNA sequences complementary to thousands of genes arrayed at precise locations |
qRT-PCR | Method for evaluating quantitative RNA expression in RNA extracted from specimens, including degraded RNA extracted from formalin fixed parafin-embedded (FFPE) tissues |
“Deep” or “next-generation” sequencing | High-throughput methods used to characterize DNA sequences, including mutations and other alternations |
| Regulation of gene expression assays | |
CLIA | Clinical Laboratory Improvement Amendments—regulations which cover approval of diagnostic tests, including multiparameter assays |
IVDMIA | In vitro diagnostic multivariate index assay—term used by the United States Food and Drug Administration (FDA) to describe certain types of multiparameter assays which are regulated as medical devices |
510(k) clearance | Regulatory approval by US Food and Drug Administration (FDA) for medical devices characterized as an IVDMIA |
| Standards for analyzing and refforting gene expression data | |
MAQC | Microarray Quality Control—effort initiated by US FDA to standardize methods for clinical application of microarray and other genomic assays |
REMARK Guidelines | “REporting recommendations for tumor MARKer prognostic studies”—standard criteria for reporting publications about tumor markers, including multiparameter gene expression assays |
MIAME | “Minimal information about a microarray experiment”—set of standards for release of gene expression data |
GEO | “Genomic Expression Omnibus” —(http://www.ncbi.nlm.nih.gov/ geo)—publicly available repository for gene expression data |
 GENOMIC INSTABILITY
GENOMIC INSTABILITY
Genomic DNA is commonly exposed to DNA-damaging agents (e.g., chemical mutagens, radiation) (17).Genomic integrity is maintained by the cellular machinery, which controls chromosomal segregation during cell division, but also by an elaborate network of genomic surveillance mechanisms and DNA repair pathways. These mechanisms are described in Table 1 and include nonhomologus end joining, homologous recombination, base excision repair, nucleotide excision repair, mismatch repair, and trans-lesion synthesis (18). Genomic instability due to heritable mutations in DNA repair genes is associated with an increased risk of colorectal, breast, and other cancers. For example, hereditary nonpolyposis colon cancer (Lynch syndrome) is characterized by mutations in DNA mismatch repair genes (mainly hMLH1 or hMSH2, less frequently hMSH6, and rarely hPMS2) which lead to genetic instability in small repetitive DNA regions, called microsatellites (spanning 1–5 base pairs, repeated 15–30 times) (17, 18). Although the precise function of microsatellites remains to be fully determined, modifications of these sequences are associated with increased risk of cancer. In the case of hereditary breast cancers, identification of germline mutations in a variety of genes results in genomic instability and increased breast cancer risk, including breast cancer susceptibility associated 1 (BRCA1) and 2 (BRCA2) genes, partner and localizer of BRCA2 (PALB2), BRCA1 interacting protein 1 (BRIP1), Checkpoint kinase 2 (CHEK2—associated with arrest of cell cycle after DNA damage), RAD51, and other genes coding for proteins involved in DNA repair or cell cycle checkpoints (19–23).
Genomic Instability in Sporadic Breast Cancers
Identification of mutant genes in molecular pathways involved in the maintenance of DNA and repair of DNA damages, in hereditary breast cancers and other types of hereditary cancers, strongly supports the “mutator phenotype” hypothesis (24, 25). This hypothesis states that genomic instability present in precancerous lesions (single cells, clonal cells, or cancer stem cells) drives tumor development by increasing the rate of spontaneous mutations (Figure 1). Three scenarios have been proposed to explain spontaneous mutations, including (a) sequential and uniform order of mutations in cancer-associated genes, where each mutation confers cellular advantages to cancer cells; (b) successive waves of clonal selection; and (c) multiple mutations in multiple pathways, in which tumor cells evolve as a heterogeneous collection. Genomic research in sporadic breast cancers has focused on identifying “caretaker genes” which are associated with microsatellite instability (MSI), chromosomal instability (CIN), and base-pair mutations (26, 27).
Microsatellite Instability
In sporadic breast cancers, several studies have evaluated microsatellite polymorphisms and mutations in mismatch repair genes which may correlate with the development of breast cancer (28, 29). Considering that several genes are involved directly and indirectly in mismatch repair, and that point mutations or epigenomic changes in their expression may affect their eficiency, studies performed to date have not reached a consensus on the relationship between breast cancer and MSI (30–32). Using polymerase chain reaction (PCR) to evaluate known polymorphism in mismatch repair genes (MSH3, MSH4, MSH6, MLH1, MLH3, PMS1, and MUTYH) in 287 breast tumor samples, Conde et al. (30) suggested that potential variants of MSH3 and MSH4 may correlate with risk of breast cancer. Although some studies have shown that mutations in mismatch genes may be associated with the development of sporadic breast cancers, it has been suggested that they may arise as secondary random events in breast cancer progression with MSI as a later repercussion on the genome.
Chromosomal Instability
Whereas MSI is characterized by changes in short DNA repeat sequences, CIN is characterized by the high rate of gain or loss of whole chromosomes or fractions of chromosomes in cancer cells in comparison with normal cells. CIN can be characterized by chromosome mis-segregation (during cellular division) and/or by structural rearrangements of chromosomes (translocations, deletions, inversions) that may modify the number of chromosomes detected in breast cancer cells resulting in aneuploidy and loss of heterozygosity (LOH) (33). Measurements of chromosome mis-segregation rates in CIN cancer cell lines have shown that these cells mis-segregate a chromosome, on average, once every one to five cell divisions (34). CIN positively correlates with cancer progression and poor patient prognosis, indicating that increased genetic diversity may be a contributing factor (33).
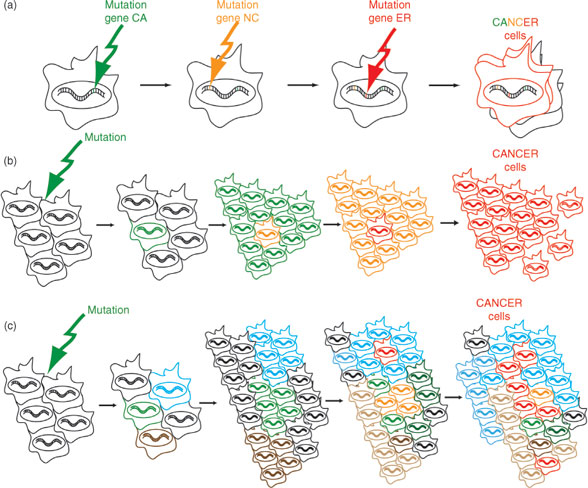
FIGURE 1
Different hypotheses on the implication of mutations for generation of cancer cells: (a) A sequential order of mutations in cancer associated genes (hypothetical names CA, NC, and ER genes) that lead to the formation of CANCER cells (red cells). (b) Successive waves of clonal mutations (original cells in black) progressively (successive mutations characterized by green, orange, and red cells) giving rise to cancer cells (red cells). (c) Multiple mutations (original cells in black), in multiple pathways giving rise to heterogeneous population of cells (characterized by blue, green, and brown cells), and ultimately giving rise to cancer cells (red cells).
 MOLECULAR TOOLS FOR EVALUATING THE GENOME
MOLECULAR TOOLS FOR EVALUATING THE GENOME
Genomic aberrations are critical events in carcino-genesis, and research efforts have been directed toward global approaches to characterize and catalogue changes that occur in human breast cancer and other types of cancers. Methods available for characterizing genetic variation in tumors include gene expression profiling, comparative genomic hybridization (CGH), single nucleotide polymorphisms (SNPs), and genomic sequencing.
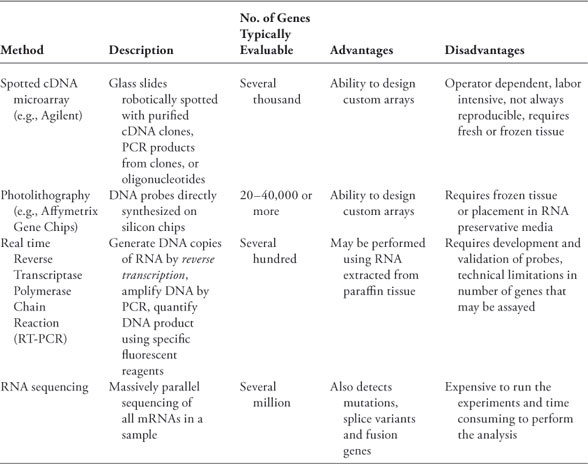
There are several methods available for analyzing gene expression which have been reviewed extensively elsewhere (summarized in Table 2 and illustrated in Figure 2), including microarrays, photolithography, quantitative reverse transcriptase polymerase chain reaction (qRT-PCR), and “deep sequencing.” For all methods, mRNA is first extracted from the tissues of interest. Because mRNA is highly vulnerable to degradation, sample handling is critical; surgical specimens should be frozen as soon as possible, or placed in appropriate preservative (e.g., RNA Later). Following mRNA lubrication, several platforms can be used to perform gene expression profiling. Prior to statistical analysis, the data must be normalized to compensate for variation in labeling, hybridization, and putrescent detection, and filtered using specific criteria to reduce the likelihood of detecting noise. Data are often summarized by a heatmap, in which patient samples are typically represented in columns, and genes in rows. Genes that are expressed at levels above the median (or a reference standard) are colored in red, close to the median in black, and below the median in green. Other color schemes are also commonly used (e.g., blue-yellow) which may be especially helpful for individuals with red-green color blindness. By comparing the expression level of a large number of genes in each of the samples, the technique of hierarchical clustering can be used to determine which samples are most similar in gene expression.
TABLE 2
Commonly used methods for measuring gene expression
There are several steps typically involved in developing a gene expression signature, including identifying the signature in a training set, and then validation of the signature in other data sets. There are methods for internally cross-validating the signature in the same data set, but external validation is always necessary in other datasets. Failure to properly adhere to these fundamental principles has led some to challenge the validity and reproducibility of microarray-based studies (35). Criteria have been developed for assessing and reporting tumor markers, including multiparameter gene expression assays, called the REMARK guidelines (REporting recommendations for tumor MARKer prognostic studies). The guidelines provide criteria for proper study design, prespecified hypotheses, assay methods, and statistical analysis methods. Standards termed “minimal information about a microarray experiment” (MIAME) for reporting the data have been established by the Microarray Gene Expression Data Society (36). Most journals require adherence to the REMARK and MIAME standards, and depositing the gene expression data in a publicly available data base (e.g., Gene Expression Omnibus) (http://www.ncbi.nlm.nih. gov/geo).
Approval of multiparameter breast cancer assays had been regulated in the past under the provisions of the Clinical Laboratory Improvement Act of 1988, which provided the basis for approval of the Oncotype DX assay (37). The regulations apply to laboratories that examine human specimens for the diagnosis, prevention, or treatment of any disease or assessment of health. The US Food and Drug Administration released a guidance document in 2007 indicating that approval of multiparameter assays falls under regulatory jurisdiction of the agency under regulations governing approval of medical devices (http://www.fda.gov/ MedicalDevices/DeviceRegulationandGuidance/ GuidanceDocuments/ucm079163.htm#1). A gene expression profilingtest system for breast cancer prognosis was defined as a device that measures the RNA expression level of multiple genes and combines this information to yield a signature (pattern or classifier or index) to aid in prognosis. Approval provided by this mechanism is commonly known as “510(k)” clearance.
Genomic (“Deep”) Sequencing
The key differences between gene expression profiling and genomic sequencing are shown in Table 2 and illustrated in Figure 3. These differences include not only the ability to evaluate expression of the entire genome, but also the ability to identify mutations and other gene variations. The Human Genome Project (http://www.ornl.gov/ sci/techresources/Human_Genome/home.shtml) used the automated Sanger sequencing method to map 3 billion nucleotides which constitute human DNA and identify about 25,000 human genes, including many genes whose functions remain to be determined. After the project was completed in 2003 using “first-generation” sequencing technology, faster and more economical methods, referred to as next-generation sequencing (NGS), have been developed (38). There are currently two major approaches to perform NGS, as described later and illustrated in Figure 3.
Sequencing After Amplification of Template Originating from a Single DNA Molecule
In principle, DNA molecules originating from a small population of cells (as little as 100 cells) are fragmented (38, 39). Specific nucleotides also defined as primers (known as synthetic nucleotidic sequences) are ligated onto the extremities of the DNA molecules and used for initiation of amplification processes. The amplification can be performed by emulsion PCR (emPCR) or by solid-phase amplification. For emPCR, the DNA is amplified, to create a DNA library, prior to binding onto the platforms where it will be analyzed. For solid phase amplification, the primers ligated on the DNA molecules allow hybridization to complementary sequences immobilized on the platform, and the amplification is performed directly on the solid platform.
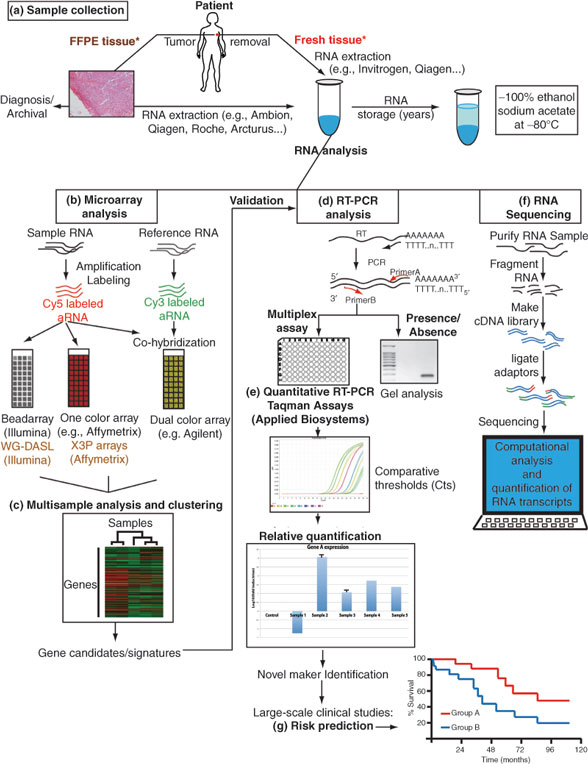
FIGURE 2
Sequencing from Single DNA Templates
Although PCR or solid phase amplification methods offer advantages for initial DNA input, they may have the disadvantage of introducing mutations during amplification processes and the potential to bias results by preferentially amplifying AT-rich compared with GC-rich DNA sequences.To date, single DNA template sequencing can be performed using three methods: (a) by ligating primers onto the fragmented DNA and hybridizing them on solid platforms where complementary primers are covalently bound; (b) by covalently binding the fragmented DNA onto a platform; and(c) by using evenly immobilized DNA polymerases to copy the primed DNA molecules.
The application of NGS has already been successful at mapping point mutations, genomic rearrangements, and changes in gene-copy number in the genomic DNA of lung carcinoma cell lines, breast carcinoma cell lines, and primary breast cancer cells (40, 41). To date, these studies have provided the most comprehensive insight into patterns of somatic rearrangement in cancer genomes, while showing that, in breast cancer, most rearrangements are intrachromosomal. Interestingly, many novel in-frame fusion genes or internally rearranged genes that were identified were expressed, which provided novel targets for research. It is suggested that multiple, infrequently rearranged cancer genes are operative in breast cancer, similarly to what has been observed in other cancers and more specifically leukemia (42–44). These studies demonstrate that the sequencing of large numbers of cancer genomes may help catalogue all classes of somatic mutations and provide insights into the molecular basis and progression patterns of cancers. Sequencing studies that potentially allow the identification of MSI, CIN, and point mutations suggest that this technology may become part of the clinician’s tools for diagnosing, treating, and predicting the clinical outcomes of their patients with breast cancer. It is important to note that the identification of “driver” mutations, which are the ones directing the process of carcinogen-esis, may be dificult to difierentiate from “passenger” mutations, which are mutations that do not participate in carcinogenesis, and may require elaborate multidimensional studies for functional validations.
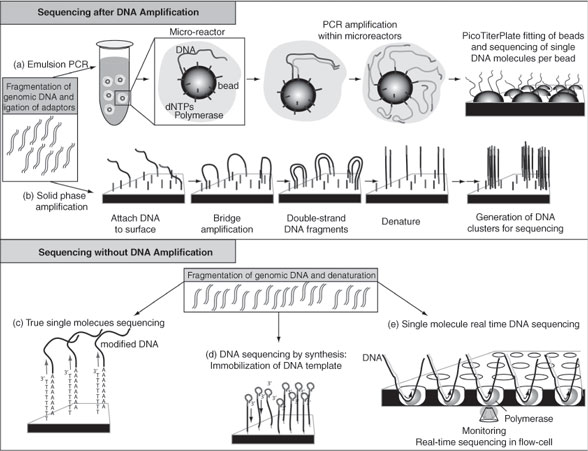
FIGURE 3
Comparative Genomic Hybridization
CGH has improved the rate of discovery by providing an eficient approach to scan entire genomes for variations in DNA copy numbers (losses, deletions, gains, amplifications) (Figure 4). In CGH measurements, genomic DNA from a test and a reference population of cells are differentially labeled and hybridized to a representation of the genome on the array. The binding of sequences, from test compared with the reference genomic DNA, provides a qualitative measure for identification of chromosomal rearrangements (45). Bacterial artificial chromosomes (BAC), complementary DNA, and oligonucleotide spotted microarrays represent technological advances for the analysis of genomic DNA copy number, and have been termed array CGH approaches. Using BAC array CGH to explore genome copy number changes in 145 human primary breast tumors, Chin et al. determined that although the genomes of clinically similar breast cancers appeared remarkably different, some regions of the genome were recurrently aberrant (46, 47). By studying the correlation between genome copy number changes and gene expression profilesof these same tumors, they identified that 10% to 15% of the entire genome was deregulated by recurrent genome copy number abnormalities. These CGH measurements suggest the existence of three general breast cancer classes, simple genomes with a few changes, complex genomes with a large number of chromosomal changes, and genomes with high-level amplifications. Experiments by Chin et al. (46) showed that tumors with BCRA1 mutations display complex genomic rearrangements, suggesting that different DNA repair pathways may be affected differently in tumors with simple, complex, or high-level amplification genomes. Studies performed with array CGH on clinical samples have also suggested that the coamplification of certain chromosomal regions 8p11–8p12 and 11q12–11q14 or 8q24 and 17q21 that contain known oncogenes and tumor suppressor genes(i.e., c-myc and HER-2) may contribute collectively to breast cancer pathophysiology (47, 48). Studies on MCF7 breast cancer cells also suggest that chromosomal rearrangements may contribute to the expression of chimeric mRNA transcripts, also known as fusion genes, which may provide or suppress molecular mechanisms that support carcinogenic events (43, 49). These studies demonstrate the necessity of mapping the DNA of tumor cells and identifying putative genes involved in the carcinogenetic events in deferent breast cancer subtypes.
SNP Arrays
SNPs, estimated to occur every 300 nucleotides, represent another level of genetic variability. It is estimated that there are more than 10 millions of SNPs in the human genome. The international effort, named HapMap (http://hapmap. ncbi.nlm.nih.gov/), initiated in 2002, has aimed at determining the common patterns of DNA sequence variations in the human genome and making it available publicly. Results obtained from the HapMap project have provided sequence information necessary for the synthesis of SNPs arrays (50, 51). It has been shown that SNPs of drug or toxicant metabolizing genes influence susceptibility of an individual to breast cancer (52, 53). SNPs in cytochrome P-450 (CYP) 1A1, CYP2A1, CYP2B6, CYP2C, CYP2D6, CYP3A, glutathione-S-transferase (GST) M1, GSTT1, GSTP1, sulfotransferase (SULT) 1A1, SULT1A2, uridine diphosphateglucuronosyltransferase, andmethylenetetrahydrofolate reductase (MTHFR) genes have been studied for their potential association with breast cancer risk and treatment outcomes in many populations (53). Although an association between changes in the SNPs of certain xenobiotic metabolizing genes and certain types of breast cancers have been reported, the use of SNPs as a predictive tool remains uncertain.
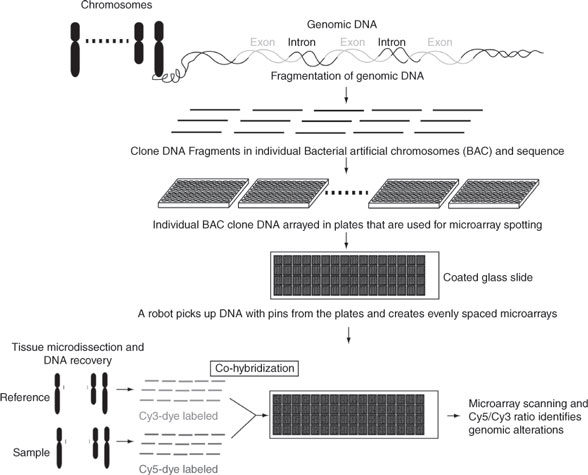
FIGURE 4
Comparative genomic hybridization can identify genomic alterations: Genomic DNA is enzymatically fragmented and the fragments are cloned into BAC. Individual BAC clones are grown into bacteria, purified, and digested to recover large amounts of each original genomic DNA fragments from the entire the genome. Small amounts of each individual DNA fragments are then spotted onto glass slides to generate DNA arrays. Genomic DNA recovered from a sample and a reference are labeled using cy5-dye and cy3-dyes, and co-hybridized onto the array. The Cy5/Cy3 intensity ratio allows for identification of genomic regions that are altered.
 EPIGENETIC REGULATION OF GENE EXPRESSION
EPIGENETIC REGULATION OF GENE EXPRESSION
Historically, the word “epigenetics” was used to describe cellular events that could not be explained by genetic principles (54). Conrad Waddington (1905–1975) defined epigenetics as “the branch of biology that studies the causal interactions between genes and their products, which bring the phenotype into being.” Epigenetics involves the study of changes in the regulation of gene activity and expression that are not dependent on gene sequence, including heritable changes, and stable long-term alterations that are not necessarily heri-table. Although epigenetics refers to the study of single genes or sets of genes, epigenomics refers to more global analyses of epigenetic changes across the entire genome. Epigenetic mechanisms control stem cell differentiation and organogenesis and contribute to the biological response to endogenous and exogenous forms of stimuli that result in disease.
An interesting example of epigenomic control of gene expression in breast cancer is the overexpression of HER-2 without genomic amplification (55, 56). A study performed on 84 human primary breast tumors displayed that up to 17% of the tumors displayed increased expression of HER-2 protein expression while lacking genomic amplification, determined by fluorescent in situ hybridization (FISH) (56). This study suggests that breast cancer cells from these patients, defined as 3+ by IHC or negative by FISH, expressing higher levels of hormone receptors may regulate the expression of HER-2 through transcriptional processes. The investigators have hypothesized that estrogen receptors (ERs) may activate the expression of HER-2, as in vitro studies have shown that AP-2 transcription factors, which have binding sites in the promoter region of HER-2, can bind estrogen-activated ERs and transduce estrogen activation (57). The investigators have also suggested that these patients may benefit from hormonal therapy. Results like these demonstrate that epigenomic processes may be as important as MSI, CIN, and point mutations for deregulating the expression of genes. Epigenomic processes can be separated in two components, the ones controlling DNA structure (chromatin acetylation, phosphorylation, and methylation), and the ones determining the function of products expressed from the genomic DNA (noncoding RNA, mRNA splicing).
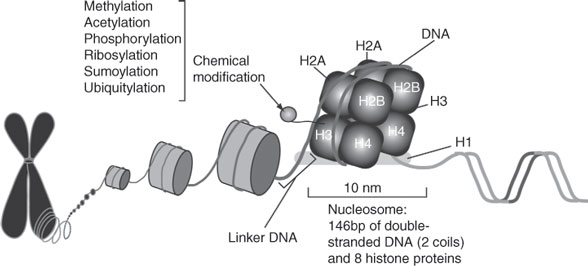
Chromatin Remodeling
Genomic DNA from eukaryotic cells is estimated to measure 1.7 m and it is efficiently packed in a structure called chromatin to fit a 5-μm nucleus (Figure 5). Chromatin’s elementary unit is called a nucleosome, which is an octamer composed of eight histone proteins and one histone protein securing the nucleosome. It is estimated that 146 nucleotides are sequentially folded around nucleosomes (~2 DNA coils per octamer) and that molecular interactions between the terminal tails of the histones and the genomic DNA can determine gene transcription, DNA repair, DNA replication, and recombination. Chemical modifications of nucleosomes can regulate “chromatin-remodeling” and include acetylation, phosphorylation, methylation, ribosylation, and SUMOylation ubiquitylation, and are coordinated by nuclear enzymes (58, 59). Chemical modifications of histones interfere with DNA compaction and promote gene expression.
Acetylation
The N-ɛ-acetylation of lysine residues found in histones is equilibrated by two enzymes: the his-tone acetyl transferases and the histone deacetylases (HDAC). There are currently 18 HDACs identified in humans that have been separated in four classes which depend on the cofactors regulating them, their functions, and their locations in human tissues. HDAC inhibitors (HDACi), which enhance histone acetylation, promote the expression of genes and have been used to promoted reexpression of silenced tumor suppressor genes. For example, the class I isoform-selective HDACi entinostat has been shown to inhibit cell proliferation and survival by inactivating downstream signaling in erbB2-overexpressing breast cancer cells that also express erbB3 (59). This occurs via downregulation of erbB3 expression and inactivation of the Akt and/ or MAPK signaling; other effects include cell cycle arrest, predominantly at the G1 phase by reduction of both cyclin D1 and the transcription factor E2F1 and upregulation of the cyclin-dependent kinase inhibitor p21(Waf1/Cip1/Sdi1/CAP20). These results suggest that specific HDACi may target specific pathways and may become useful complements in endocrine and/or growth factor inhibitor therapies for the treatment of certain breast cancer subtypes.