Highlights
- •
Small molecule MIF inhibitor CPSI-1306 inhibits HNSCC growth in vivo .
- •
CPSI-1306 potently inhibits T cell immunosuppression in the tumor microenvironment.
- •
CPSI-1306 completely abrogates CTLA4, TIGIT and TIM3 in cytotoxic CD8+ T cells.
Abstract
Head and neck squamous cell carcinoma (HNSCC) is the 7th most common cancer globally with a 40–50 % survival rate. Although macrophage migration inhibitory factor (MIF) is overexpressed in most solid tumors and promotes tumor growth and invasion, the therapeutic potential of MIF inhibition in HNSCC is yet to be explored. In this study, we investigated the efficacy of CPSI-1306, a small-molecule MIF inhibitor, on HNSCC cell growth and cancer associated signaling pathways in vitro , as well as its impact on T cells in the HNSCC tumor microenvironment in vivo . CPSI-1306 did not reduce HNSCC cell proliferation in vitro , and mildly decreased VEGF and EGFR expression. However, CPSI-1306 significantly reduced tumor development in two orthotopic mouse oral cancer (MOC-2 and MOC-1) HNSCC models. Interestingly, CPSI-1306 treatment increased T cell infiltration to the tumor microenvironment and completely abrogated immunosuppressive checkpoint markers TIGIT, TIM3, and CTLA-4, but not PD-1 on tumor infiltrating CD8 + T cells. This was accompanied by increased CD8 + T cell expression of antitumoral cytokines IFN-γ and TNF-α in the draining lymph nodes and Granzyme B in the tumor microenvironment of CPSI-1306 treated tumor bearing mice. Our studies demonstrate that the small molecule MIF inhibitor CPSI-1306 potently inhibits T cell immunosuppression in the tumor microenvironment and reduces tumor growth in HNSCC. These studies open a novel therapeutic option for modulating anti-tumoral T cell immunity to improve HNSCC outcomes by targeting MIF.
Introduction
Head and Neck Squamous Cell Carcinoma (HNSCC) is the 7th most common cancer worldwide with a 5-year survival rate of 40–50 % . Human Papilloma Virus (HPV) negative HNSCC, partly attributable to alcohol and tobacco consumption, is associated with a worse prognosis . Treatment for HPV negative HNSCC consists of radiation and highly invasive surgery which causes disfigurement. Chemoradiotherapy, with cisplatin/docetaxel followed by radiation has increased progression free survival in recent clinical studies . However, chemoradiotherapy related toxicities have become a growing concern with neutropenia, thrombocytopenia, and dysphagia, ruling out elderly patients and those with comorbidities due to treatment related deaths . Recently, molecular and immune therapies have emerged as viable treatment options, with multi-modal approaches including anti-PD-1 antibody, VEGF and mTOR inhibitors, and EGFR inhibitors such as cetuximab demonstrating modest increases in median overall survival when combined with chemotherapy . Neoadjuvant immunotherapy such as PD-1 checkpoint blockade is emerging as a promising strategy for improving anti-tumor immune responses, particularly in head and neck squamous cell carcinoma (HNSCC). Although PD-1 checkpoint blockade has been approved as first line treatment for advanced HNSCC patients, underlying mechanisms of enhanced treatment responses, as well as optimal therapeutic regimens for improved disease and progression free survival are still incompletely understood .
Studies investigating factors affecting efficacy of neoadjuvant therapy demonstrate that systemic immune responses play a central role in tumor eradication. For example, Spitzer et al showed that peripheral immune activation, particularly the expansion of CD4 + T cells, is essential for the long-term protection against tumor recurrence and for generating immune memory . More recent studies highlight the critical role of lymph nodes in orchestrating immune responses during PD-L1 immunotherapy in HNSCC patients. One such study demonstrated that progenitor exhausted CD8 + T cells in uninvolved lymph nodes are clonally related to exhausted T cells in the tumor, and that anti-PD-L1 therapy can promote their activation and differentiation . Together, these studies highlight that effective neoadjuvant immunotherapy not only targets the tumor microenvironment but also induces broad systemic immune activation, with lymph nodes acting as critical hubs for immune activation and coordination across tissues. These insights suggest that combining local and systemic immune responses could significantly enhance therapeutic outcomes in HNSCC and other cancers. However, there remains a great need for further research into therapeutic options that target the HNSCC tumor microenvironment to enhance the responses to these immunotherapy treatments and improve anti-tumoral efficacy.
Macrophage migration inhibitory factor (MIF) is a pleiotropic cytokine produced by immune and non-immune cells, originally discovered in the 1960s to inhibit the random migration of macrophages . MIF is involved in regulating innate immunity, as it can augment expression of various cytokines including TNF-α, IL-1, IL-6, and IL-8. MIF also counteracts the immunosuppressive effects of glucocorticoids . Numerous studies since then have identified MIF as an important biomarker of cancer progression, being overexpressed in multiple solid tumors. MIF has been associated with osteosarcoma, gastric cancer, lung squamous cell carcinoma, pancreatic cancer, and various forms of HNSCC including nasopharyngeal, oral squamous cell, and esophageal squamous cell carcinomas . MIF binds to CD74/CD44, CXCR2, or CXCR4, and promotes activation of cancer associated MAPK and PI3K-AKT pathways related to chronic inflammation, proliferation, and apoptotic evasion . MIF also promotes the expression of biomarkers of cell invasion and metastases, and MIF knockdown downregulates expression of matrix metalloproteins MMP2/9 . Importantly, MIF expression is correlated with cancer aggressiveness . As such, targeting MIF potentially offers a viable approach in the treatment of solid tumors.
The mechanistic role of MIF in HNSCC is still under active investigation. Early reports demonstrated that exogenous MIF promotes the invasive phenotype of HNSCC cells . High MIF expression in HNSCC tumor tissue is linked to recurrence, metastasis, and poor survival, and MIF knockdown in HNSCC cells decreases cell proliferation and migration, potentially mediated by modulation of MAPK pathways . Previous research by our group further demonstrated that genetic MIF deletion results in improved HNSCC outcomes, associated with decreased inflammatory markers, including IL-1β, TNF-α, and chemokines CCL3 and CXCL1 . This is further supported by Kindt et al , where mice with decreased MIF expression are reported to have better survival, delayed onset of tumors, and better responses to chemotherapy . Recently, Chen et al. demonstrated that MIF knockdown can be combined with UV radiation therapy to promote HNSCC anti-cancer effects . These studies provide insights on the mechanistic effects of MIF on cancer associated pathways, and immunosuppression in the HNSCC tumor microenvironment. Further, these studies highlight the need for in-depth investigation of non-toxic, molecular MIF inhibitors in HNSCC treatment.
MIF inhibition via anti-MIF antibodies was a preclinical option for targeting MIF in the late 1990s and 2000s . However, in the past few decades targeting MIF via small molecule inhibitors has proven to be a more effective, better tolerated, and cost-effective approach to cancer treatment . Small molecule MIF inhibition offers a direct way of blocking MIF binding to its corresponding ligands, via interaction with its tautomerase binding site. Various classes of small inhibitors have shown effectiveness in targeting MIF in thyroid cancers, squamous cell carcinoma, glioblastoma, and colon cancers. 4-IPP, a phenylpyrimidine class of inhibitors, has shown significant pharmacological inhibition of MIF in squamous cell carcinoma, decreasing cell proliferation on a dose-dependent basis . ISO-1, a newer generation isooxazoline class inhibitor, has been implicated in inhibiting MIF and reducing cell growth in melanoma, prostate, and colon cancers . Interestingly, Wang et al reported that CPSI-1306 may be up to 100 times more potent when compared to ISO-1, providing a greater indication of using this newer line of inhibitors as a potential therapeutic for targeting cancer. CPSI-1306, is an isoxazoline class of MIF inhibitor that has shown significant results in murine models of triple negative breast cancer and UVB induced squamous cell carcinoma but has not been studied in head and neck cancer . CPSI-1306 inhibits the trimerization of MIF secreted by cancer cells, decreasing its ability to bind to its respective ligands and receptors and decreasing its pro-cancer effects. Given the greater potency of MIF inhibition by CPSI-1306 compared to previous generation MIF inhibitors, combined with its reported minimal toxicity to normal cells, CPSI-1306 offers a potentially viable approach to targeting MIF associated pathways in HNSCC treatment.
In this study we determined the therapeutic efficacy of MIF inhibition with CPSI-1306 in HNSCC treatment using in vitro and two in vivo models (MOC-2 and MOC-1). We further describe mechanisms underlying CPSI-1306 mediated inhibition of oral carcinogenesis, specifically via inhibition of T cell immunosuppressive pathways in the HNSCC tumor microenvironment. Our results reveal a novel therapeutic option for modulating anti-tumoral T cell immunity to improve HNSCC outcomes by targeting MIF.
Materials and methods
Cell culture and treatments
CPSI 1306 was generously provided by L2 Diagnostics, LLC New Haven, CT. HNSCC cell lines CAL-27 SCC-83 and CA-83, were cultured in DMEM, 10 % FBS, and 1 % Pen-strep glutamine with supplemental 1 % non-essential amino acids. MOC2 cells were cultured in IMDM/F12, 2:1, with 5 % FBS, 1 % Pen-strep glutamine, 5 ug/mL insulin, 40 ng/mL hydrocortisone, and 5 ng/mL human recombinant EGF.
Cell viability assay
Cell viability assay was performed using alamarBlue dye (Thermo Fisher Scientific). 2 × 10 3 cells were seeded per well in 100 μl of medium into 96-well plates. Cells were allowed to incubate overnight and serially treated with 100 µM with CPSI-1306 for 72 h. After incubation, 10 μl alamarBlue dye was added and incubated for 6 h. Absorbance was measured at 570 nm and 600 nm, and percent viability was calculated as described.
Histology
Tumors were formalin-fixed and paraffin-embedded as previously described . For immunohistochemistry (IHC), Tissue sections (5 µm) were stained for MIF (Proteintech, 20415-1-AP) with secondary biotinylated antibody for goat anti-rabbit (Vector Labs, BA-1000) along with a hematoxylin counterstain. For immunofluorescence (IF), tissue sections (5 µm) were stained for CD8 (Invitrogen, A21434) and Granzyme B (Abcam, AB4059) with the secondary antibodies Alexa Fluor 488-conjugated goat anti-rabbit (Invitrogen, A11034) and Alexa Fluor 555 goat anti-rat (Invitrogen, A21434). Each section was then counterstained with DAPI (BioLegend, San Diego, CA). Confocal imaging was performed sing a Zeiss LSM 700 confocal microscope (CarlZeiss, Munich, Germany). Regions of positive stains were quantified using ImageJ for both IHC and IF.
Real time quantitative PCR
Human and mouse primer sequences for BCL2, VEGFA, EGFR and CCND1 were obtained using PRIMER BANK ( https://pga.mgh.harvard.edu/primerbank/index.html ). PCR amplification was performed using the PowerUp SYBR Green Master Mix (Thermofisher Scientific, Foster Coty, CA, USA) for detection. Data normalization was performed to housekeeping genes GAPDH and β-actin.
Western blotting
Proteins were extracted from in vitro cultures of CAL27, SCC83, CA83, and MOC2 treated with CPSI-1306 at 1 and 10 µM concentrations for 24 h. 30 μg of protein was loaded onto a 10 % Tris-HCL gel. Proteins were transferred to a PVDF membrane, blocked using 5 % non-fat dry milk and incubated with the rabbit phospho-p44/42 MAPK (Erk1/2) (Cell Signaling, 4370S), p44/42 MAPK (Erk1/2) (Cell Signaling, 4695S) and Rabbit GAPDH (Cell Signaling, 2118S) primary antibody overnight. Blots were incubated with goat anti-rabbit HRP linked secondary antibody (Thermo Fisher Scientific, Rockland, IL). Chemiluminescence was detected by ECL western blotting substrate (Thermo Scientific, Waltman, MA). Quantification of images was performed using the ImageJ FIJI package (Ver. 2.1.0) in ImageJ.
Animals and ethics
Experiments were approved by the Institutional Animal Care and Use Committee of the Ohio State University (IACUC) and animals housed and cared for by University Laboratory Animal Resources guidelines. Female C57BL/6 mice were purchased from Jackson Laboratory (Bar Harbor, ME) and were injected orthotopically with MOC-2 (3 × 10 4 cells) or MOC-1 (5 × 10 5 cells) into the right buccal cavity. Mice were randomly assigned into vehicle control (n = 10) and CPSI-1306 (n = 10) groups after injection for each orthotopic cell line model. On days 5 and 8 post-injection, mice began treatment via oral gavage with CPSI-1306 (20 mg/kg in 15 % DMSO, 0.5 % methylcellulose in water) or vehicle 5 times per week for 2 weeks in MOC-2 tumor-bearing mice and 6 weeks for MOC-1 tumor-bearing mice. Mice weight and tumor volume measurements were recorded twice a week. Tumors were analyzed for volume via equation V = L × S 2 × 0.5, where L is longest diameter and S is short diameter. At terminal sacrifice, spleen, tumors, and lymph nodes were harvested for flow cytometry and gene expression analysis. Tumors were imaged and analyzed for volume at sacrifice.
Flow cytometry
For in vitro analysis of MIF expression, TE1177, SCC83, CA83, and CAL27 cells were incubated with MIF conjugated antibody (Invitrogen, 367401) and MOC2 cells were incubated with MIF fluorochrome conjugated antibody (ProteinTech, 20415-1-AP). Single cell suspensions were generated from spleens, draining lymph nodes and tumors of experimental mice, which were stained extracellularly with fluorochrome conjugated antibodies for CD4, CD8, CD45, CTLA4, TIM3, TIGIT, LAG3, CD69, and PD1 (BioLegend. San Jose, CA, USA). Cells were also incubated with antibodies targeting IL-2, Il-10, IFN- γ, Perforin, Granzyme B, and TNF- α to identify intracellularly expressed markers (BioLegend. San Jose, CA, USA). For myeloid subsets, cells were incubated with fluorochrome conjugated antibodies for Cd11b, CD11c, IA/IE, CD80, F4/80, Ly6C, Ly6G, and PD-L1 (BioLegend. San Jose, CA, USA). Samples were analyzed using FACS Celesta flow cytometer (BD Biosciences, San Jose, CA). Flow cytometric analysis was completed using FlowJo (Tree Star Inc., Ashland, OR, USA).
Enzyme-linked immunosorbent assay (ELISA)
Serum was collected from blood of MOC-2 and MOC-1 tumor bearing mice treated with CPSI-1306 or vehicle control. The production of cytokines IL-6 and TNF-α was measured using ELISA. All the capture and detection antibodies were purchased from BioLegend (San Diego, CA, USA).
Statistical analysis
Statistical analyses were conducted using GraphPad Prism software v9.2.0 (GraphPad Software, San Diego, CA, USA). Student’s t test (two-sided) was used to determine the statistical significance between the groups.
Results
CPSI-1306 reduces tumor progression in orthotopic in vivo models of HNSCC
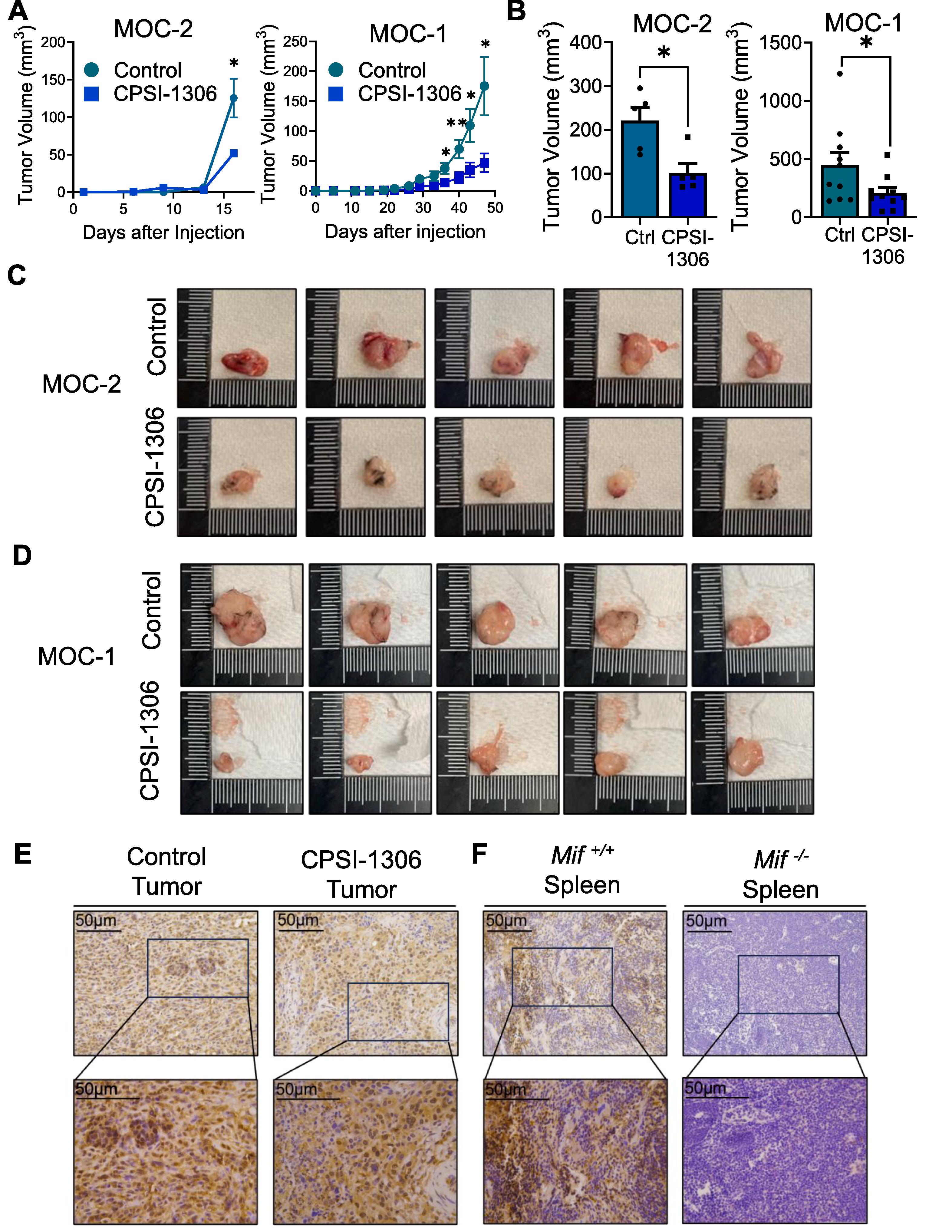
We determined the therapeutic potential of CPSI-1306 in orthotopic models of HNSCC in vivo using the well characterized MOC-2 and MOC-1 HNSCC cell lines . MOC-2 tumor bearing mice were treated with either CPSI-1306 (20 mg/kg/day for 2 weeks) or vehicle via oral gavage, 5 days following tumor injections. MOC-1 tumor bearing mice were treated with either CPSI-1306 (20 mg/kg/day for 6 weeks) or vehicle via oral gavage, 8 days following tumor injections. Mouse tumors were measured, with reduced tumor volumes noted for MOC-2 and MOC-1 tumors at Day 17 and beginning at Day 36 post injection respectively ( Fig. 1 A). Mouse weights were recorded, with no significant weight differences but on average being higher with CPSI-1306 treatment ( Supplementary Fig. 1 ). Mouse tumors were also measured and quantified using ImageJ post sacrifice, with volumes similarly being significantly reduced with CPSI-1306 treatment in both tumor models ( Fig. 1 B–D). This data suggests that CPSI-1306 can reduce head and neck cancer cell growth in vivo . We further characterized expression of MIF in mice bearing tumors after injection with MOC-2 HNSCC cells. As expected, MOC-2 tumor bearing mice expressed high MIF levels, in tumors of both CPSI-1306 and vehicle treated mice ( Fig. 1 E). MIF expression was also detected in spleens of tumor bearing mice, but not on MIF knockout mice ( Fig. 1 F).
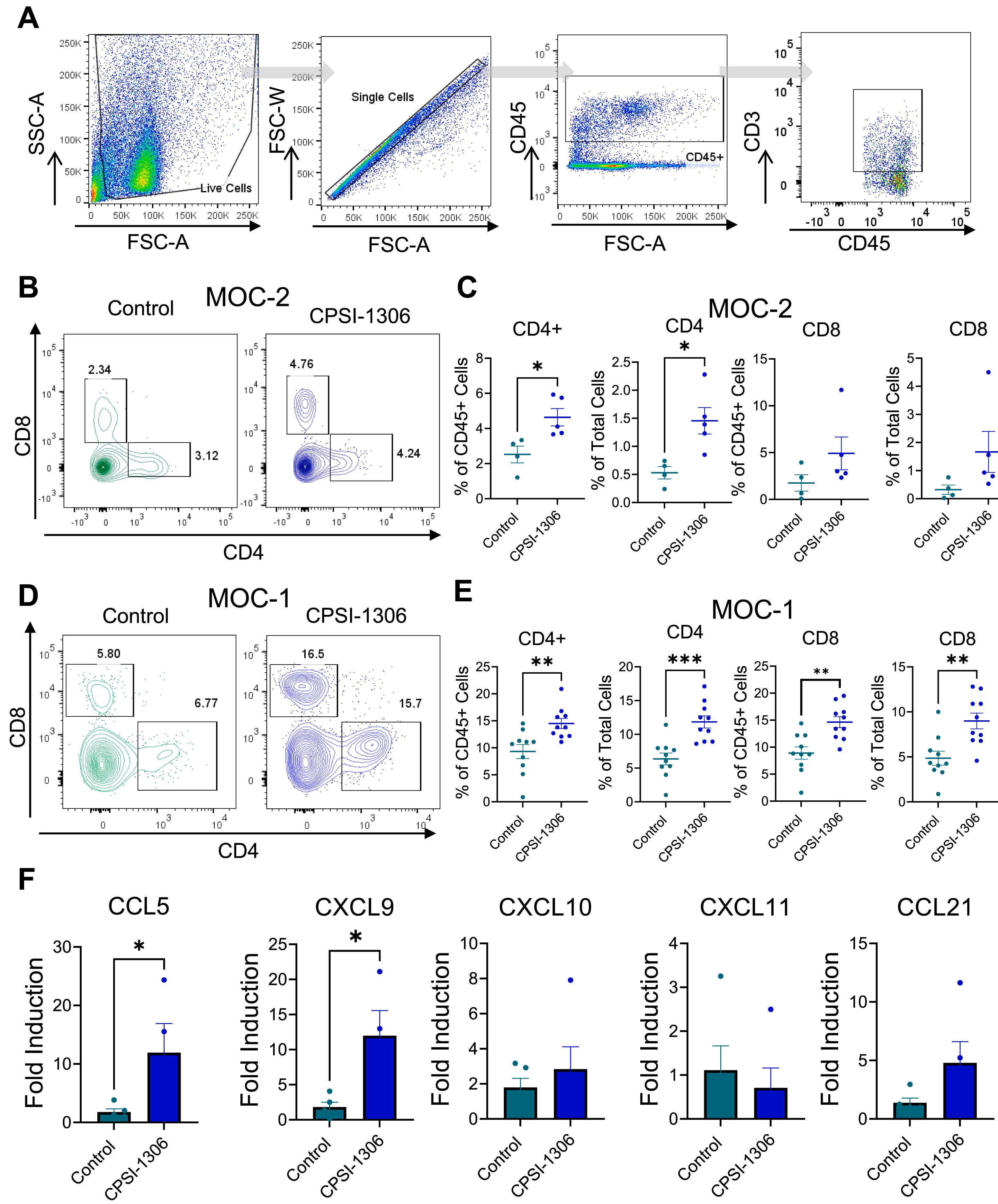
CPSI-1306 enhances T-cell infiltration to the HNSCC tumor microenvironment
We determined potential mechanisms underlying CPSI-1306 mediated anticancer effects. First, we analyzed CD4 + and CD8 + T cell infiltration into the tumor microenvironment of MOC-2 and MOC-1 tumor bearing mice ( Fig. 2 A). CD4 + T-cell infiltration was significantly increased in tumors of CPSI-1306 treated MOC-2 tumor bearing mice compared to vehicle controls, while a modest but not significant increase in CD8 + T cells was observed in CPSI-1306 treated MOC-2 tumor-bearing mice ( Fig. 2 B and C). Both CD4 + and CD8 + T cells were significantly increased in tumors of CPSI-1306 treated MOC-1 tumor bearing mice compared to vehicle controls ( Fig. 2 D and E). These findings correspond to previous studies showing that MIF inhibition may promote anticancer effects through improved immune activation and function . To determine potential mechanisms underlying the increased T cell infiltration of CPSI-1306 treated tumor bearing mice, we analyzed gene expression levels of chemokines associated with enhanced T cell recruitment. We observed increased expression levels of CCL5 and CXCL9 in the tumor microenvironment of CPSI-1306 treated MOC-2 tumor bearing mice compared to vehicle control group ( Fig. 2 C). Taken together, our data indicates that reduction in tumor progression in CPSI-1306-treated mice is associated with the upregulation of antitumoral T cell attracting chemokines and increased recruitment of T cells to the tumor microenvironment.
Related posts:
 Surgical complications and functional outcomes of 3191 jejunal free flaps used for reconstruction of circumferential defects following head and neck cancer resections: A systematic review
Surgical complications and functional outcomes of 3191 jejunal free flaps used for reconstruction of circumferential defects following head and neck cancer resections: A systematic review
 Submandibular gland transfer into the temporal fossa in patients with oral squamous cell carcinoma: A viable option to prevent radiation-induced xerostomia
Submandibular gland transfer into the temporal fossa in patients with oral squamous cell carcinoma: A viable option to prevent radiation-induced xerostomia
 Induction chemotherapy for locally advanced nasopharyngeal carcinoma: Efficacy and safety of the TPC regimen compared to GP and TPF
Induction chemotherapy for locally advanced nasopharyngeal carcinoma: Efficacy and safety of the TPC regimen compared to GP and TPF
 Predictors of recurrence and survival after salivary gland cancer surgery: A multicenter, retrospective study in northern Japan
Predictors of recurrence and survival after salivary gland cancer surgery: A multicenter, retrospective study in northern Japan
 The 10 %-rule debate: A fresh perspective on sentinel lymph node biopsy in OSCC
The 10 %-rule debate: A fresh perspective on sentinel lymph node biopsy in OSCC
 Deciphering molecular relapse and intra-tumor heterogeneity in non-metastatic resectable head and neck squamous cell carcinoma using circulating tumor DNA
Deciphering molecular relapse and intra-tumor heterogeneity in non-metastatic resectable head and neck squamous cell carcinoma using circulating tumor DNA
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


