Highlight
• Five gut microbiota were found to be causally associated with bladder cancer.
• Among these taxa, the genus Bilophila was identified as having a promoting effect on bladder cancer.
• Sensitivity analyses did not reveal significant heterogeneity or horizontal pleiotropy among the included instrumental variables.
• The gut microbiota taxa Enterobacteriaceae, Oscillibacter, Ruminococcaceae NK4A214 group, and Enterobacteriales were found to be protective against bladder cancer.
• The study suggests a potential causal relationship between genetically-based gut flora and bladder cancer, providing insights into the “bowel-bladder axis” of bacteria influencing the development of bladder cancer.
Abstract
Background
Recent studies have underscored a potential link between gut microbiota and urological tumors, yet the causal relationship with bladder cancer (BCa) and the role of metabolic pathways remain unclear.
Methods
Instrumental variables (IVs) for gut microbiota were obtained from genome-wide association studies (GWAS) conducted by the MiBioGen consortium (n = 18,340). GWAS data for BCa were sourced from a comprehensive genome-wide meta-analysis encompassing 23 cohorts. Mendelian randomization (MR) was employed to investigate the causal relationship between gut microbiota and BCa, utilizing inverse variance weighted (IVW) as the primary MR method. Additionally, metabolic pathways associated with these microbiota were analyzed to understand their functional roles in BCa pathogenesis. Sensitivity analyses were conducted to validate all MR results.
Results
The MR analysis identified five gut microbiota taxa with a causal association with BCa, with the genus Bilophila notably promoting BCa. Metabolic pathway analysis revealed significant associations between specific pathways and BCa, suggesting that changes in amino acid and NAD metabolism might influence BCa development. Sensitivity analyses indicated no significant heterogeneity or horizontal pleiotropy among the IVs.
Conclusion
This study revealed the significant causal relationship between gut microbiota and BCa, particularly identifying Bilophila as a key pathogenic initiator. These findings elucidated the potential impact of metabolic pathways, especially amino acid and NAD metabolism, on the pathogenesis of BCa. They not only laid the foundation for innovative therapeutic strategies but also highlighted the immense potential of microbiota-based interventions in the prevention and treatment of BCa, paving the way for new directions in precision medicine.
1
Introduction
Bladder cancer (BCa) is among the top 10 most common malignant tumors worldwide, with around 573,000 new cases and 213,000 deaths annually, posing a significant threat to life and health [ ]. The exact cause of BCa remains unknown, but research suggests potential links to prolonged smoking, various occupational hazards (e.g., aluminum and rubber production, exposure to industrial dyes), environmental exposures (e.g., X-ray or gamma radiation, arsenic), certain medications (such as cyclophosphamide and liver issues) [ ], as well as schistosoma infections [ ]. Additionally, correlations have been observed between BCa and factors like age, gender, and family medical history [ ]. Given the current uncertainty surrounding the mechanisms of BCa onset, exploring its underlying processes is crucial for advancing early diagnosis, treatment, and prognostic evaluation of the disease.
In recent years, there has been a growing interest in factors such as the human microbiota, organismal immunity, and regulatory immune status in relation to tumors [ ]. Recent research highlights the correlation between commensal microbes in humans and various complex diseases, including mental disorders, cardiovascular diseases, and cancer [ , ]. Koning et al. [ ] discuss how regulating intestinal flora and improving intestinal barrier function have emerged as promising therapeutic approaches for preventing and treating specific diseases. If the causal link between gut flora and tumor risk holds true, gut flora could be a potential marker for early cancer diagnosis and a better prognosis [ ]. Mendelian randomization (MR) analysis utilizes single nucleotide polymorphisms (SNPs) as instrumental variants (IVs) to investigate potential causal relationships between environmental exposures and disease [ ]. Furthermore, MR enhances the reliability of data results by avoiding reverse causality inferences and reflecting the long-term effects of exposure on outcomes [ ].
The significance of patients’ gut flora for BCa remains uncertain, with three studies conducted to date examining the gut flora characteristics of BCa patients [ ]. The available data on intestinal flora differences primarily suggest general intestinal dysbiosis rather than BCa-specific alterations. Neither study adequately addressed confounding factors nor variations in the BCa-flora association [ ]. Hence, standardized reporting and robust analyses of potential risk factors are necessary to validate the mechanisms underlying the initially observed association between BCa and gut flora. This will enhance understanding of the pathogenesis of BCa and the search for treatment and prognosis biomarkers to better inform clinical care.
2
Methods
2.1
Data sources
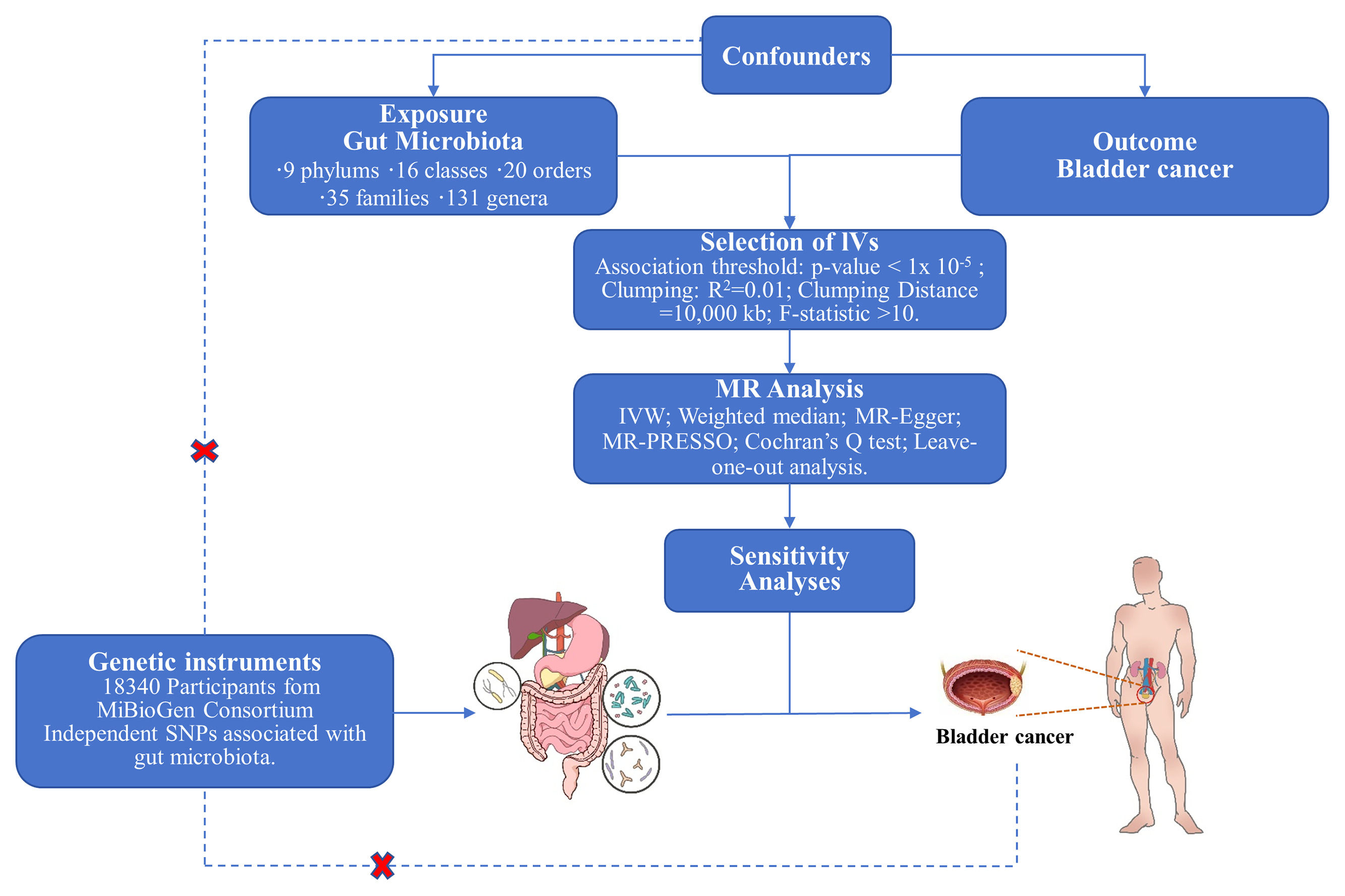
This study employs a 2-sample Mendelian randomization (MR) analysis to explore the causal link between gut flora and BCa. Data on gut flora and BCa are derived from a genome-wide association study (GWAS). The flowchart of this study is shown in Fig. 1 . Genetic variations in gut flora are sourced from the MiBioGen consortium and the Dutch Microbiome Project (DMP) on gut microbiota composition. The MiBioGen study supplied a dataset of genome-wide linked 16S rRNA gene sequencing from 24 cohorts, encompassing 18,340 participants across diverse ethnicities and age groups from Europe, Africa, and Asia, for the MR analysis of gut microbiota [ ]. 211 taxonomic units have been identified in these cohorts, including 9 phylums, 16 classes, 20 orders, 35 families, and 131 genera of associated genetic variation [ , ]. DMP possesses the largest species-level gut microbiome database to date, including metagenomic sequencing data from 7,738 participants from northern Netherlands. It has identified 207 taxa and 205 pathways, illustrating microbial composition and functionality. The GWAS catalog provides summary statistics for the metabolic pathways (entry numbers range from GCST90027446 to GCST90027857, https://dutchmicrobiomeproject.molgeniscloud.org ) [ ]. BCa-related data, involving 2,264 cases and 454,084 controls, were obtained from the public database GWAS ( https://gwas.mrcieu.ac.uk/ ), and genetic variation associated with BCa was analyzed by pooling the datasets GCST90041857 ( https://www.ebi.ac.uk/gwas/studies/GCST90041857 ) [ ]. See Table 1 .
| Variables | Consortium | Traits | Year | Population | Sample size | nSNPs | nTaxa | Websites |
|---|---|---|---|---|---|---|---|---|
| Exposure | MiBioGen | Phylum | 2021 | European | 14,306 | 9 | https://mibiogen.gcc.rug.nl/menu/main/home | |
| Class | 2021 | European | 14,306 | 16 | ||||
| Order | 2021 | European | 14,306 | 20 | ||||
| Family | 2021 | European | 14,306 | 33 | ||||
| Genus | 2021 | European | 14,306 | 119 | ||||
| DMP | – | 2022 | European | 7,738 | – | https://dutchmicrobiomeproject.molgeniscloud.org | ||
| Outcome | UK biobank | BCa | 2021 | European | 2,264 | 454,084 | – | https://www.ebi.ac.uk/gwas/studies/GCST90041857 |
2.2
The selection of instrumental variables (IVs)
To ensure the accuracy of the results of the causal effect of gut flora on BCa, appropriate SNPs were selected according to the basic principles of Mendelian randomization, and stringent quality control procedures were performed on the GWAS pooled data. Firstly, valid instrumental variables (IVs) must meet three essential assumptions: (1) IVs are strongly and reliably linked to the gut microbiota; (2) IVs are not associated with other confounding factors that might affect risk factors and outcomes; and (3) IVs can affect BCa solely through the gut flora. Second, for quality control of SNPs, we chose SNPs that were statistically significantly associated with the gut microbiome as IVs ( P < 5 × 10 −8 ). Unfortunately, only a small number of SNPs were chosen as IVs. To explore more comprehensive results, we relaxed the association p-value threshold between the instrument and the exposure, selecting SNPs with P < 1 × 10 −5 as IVs to obtain more microbial data [ ]. Consistent with previous studies, to avoid strong linkage disequilibrium (LD) that could lead to biased results, we used reference data from the 1000 Genomes Project European samples, set an LD threshold of r 2 <0.001, and set an aggregation window of 10,000 kb for our analyses [ , ]. Finally, to assess the strength of the SNPs, we calculated F statistics for each SNP to quantify the statistical strength, and F statistics <10 were excluded [ ].
2.3
Mendelian randomization (MR) analyses
To determine the correlation between intestinal flora and BCa, we used five methods: inverse variance weighting (IVW), the MR-Egger method, the weighted median method (WMe), the weighted mode method (WMo), and the simple mode method (SM). Specifically regarding the five methods under study, IVW stands out as the primary analytical approach for MR. It assumes that all IVs share a common causal effect on outcomes through risk exposure, yielding unbiased estimates of effects without horizontal pleiotropy. Unlike the MR-Egger regression, which is a method used for horizontal pleiotropy, it is assumed that pleiotropy is present in >50% of the SNPs [ ]. And, assuming polytropy in <50% of SNPs, WMe and WMo can be used as robust scenarios [ ]. After IVW confirmed a causal relationship between exposure and outcome, if the direction of the OR values of MR-Egger and WMe were in agreement with them (both: OR > 1 or OR < 1), the results were considered to be reliable, and the next step of sensitivity and specificity assessment was performed.
We used the MR-Egger and MR Polytomous RESidual Sum and Outlier (MR-PRESSO) tests for horizontal polytomousness and outliers. MR-PRESSO is divided into three parts: the global test, the outlier test, and the distortion test. Compared with MR-Egger, MR-PRESSO can more accurately identify horizontal pleiotropy and outliers and screen for abnormal SNPs [ ]. And MR-Egger was used to explain the presence of directional genetic pleiotropy; when the P -value was > 0.05, we considered that there was no statistical significance of genetic pleiotropy. Subsequently, we performed a Cochran’s Q test to test for heterogeneity between instrumental variables, and when P < 0.05, we observed heterogeneity. Finally, to infer the reliability of causal direction, we used the MR Steiger directionality test to examine whether gut flora is directionally causally related to BCa [ ].
2.4
Statistical analysis
R software (version 4.1.2) was used for all statistical and data visualization analyses in MR. MR analyses of causality between gut flora and BCa were performed using the “TwoSampleMR” package and the “MRPRESSO” package, with P < 0.05 indicating a statistically significant potential causal relationship.
3
Results
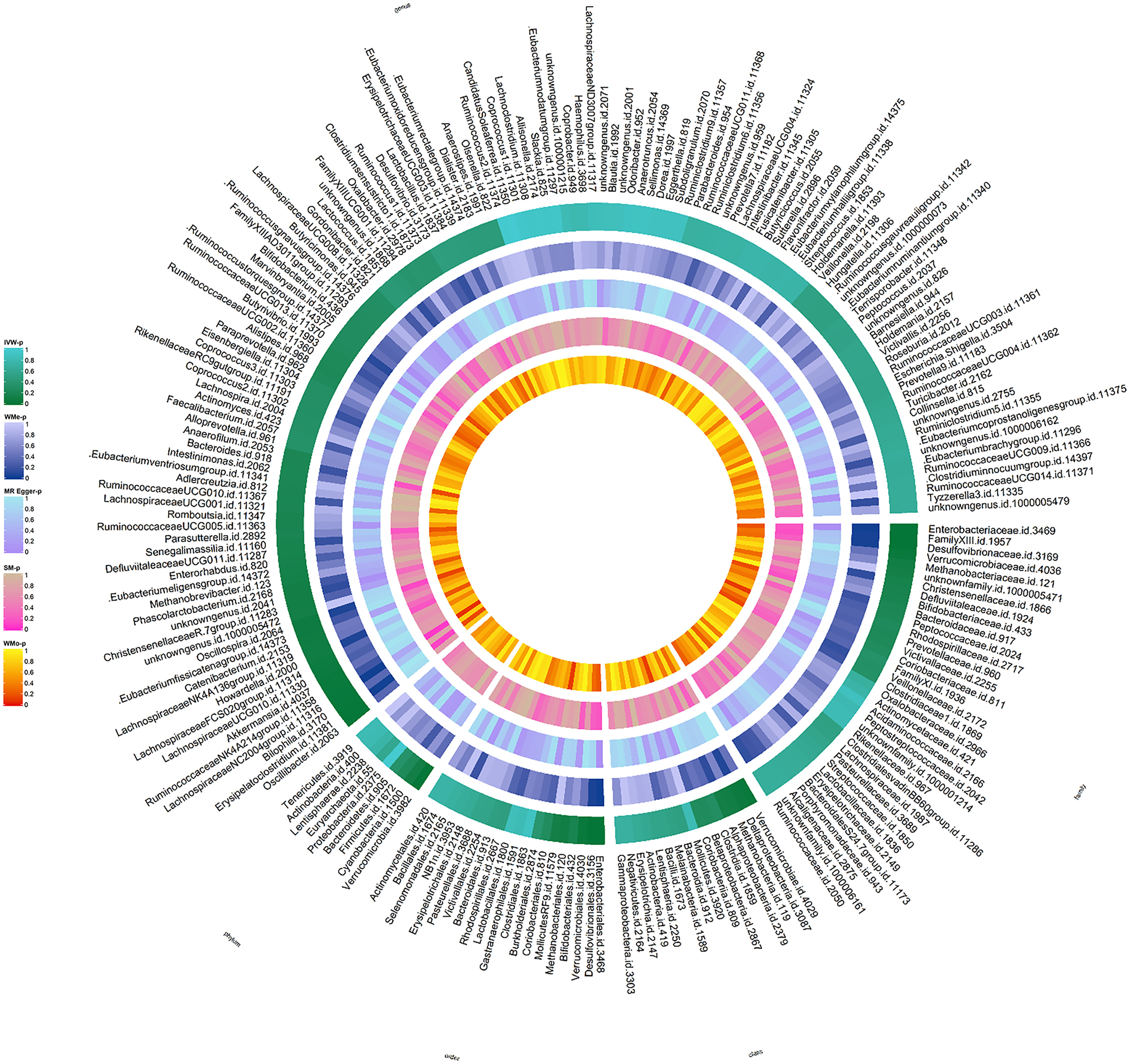
We performed MR analyses on 211 gut microbiota and screened 2,104 SNPs as instrumental variables. F statistics of all instrumental variables were calculated to obtain SNPS with F statistics > 10. The circus plot then shows the results of the MR analysis of the IVs ( Fig. 2 ). We queried the positive results for the above SNPs in PhenoScanner and found no SNPs associated with the above confounders. Finally, the results of the IVW analyses showed a significant correlation between 1 order, 1 family, and 3 genera ( Fig. 3 A), details of which are given in Supplementary Table 1 . The results of our MR Egger, weighted median analyses are shown in Supplementary Table 2 . Simultaneously, we conducted MR analysis on 205 gut microbial pathways of DMP, revealing four gut microbial functional pathways that significantly influenced BCa risk ( P < 0.05, Fig. 3 B). The details are shown in Supplementary Table 3 .
Related posts:
 Tumor involvement of the trigone and urethra at the time of robot-assisted radical cystectomy is associated with adverse oncological outcomes
Tumor involvement of the trigone and urethra at the time of robot-assisted radical cystectomy is associated with adverse oncological outcomes
 Diet and the microbiome as mediators of prostate cancer risk, progression, and therapy response
Diet and the microbiome as mediators of prostate cancer risk, progression, and therapy response
 Understanding the microbiome as a mediator of bladder cancer progression and therapeutic response
Understanding the microbiome as a mediator of bladder cancer progression and therapeutic response
 5α-reductase inhibitors with or without alpha-blockers and risk of incident upper tract urothelial carcinoma in men with benign prostatic hyperplasia: Analysis of US insurance claims data
5α-reductase inhibitors with or without alpha-blockers and risk of incident upper tract urothelial carcinoma in men with benign prostatic hyperplasia: Analysis of US insurance claims data
 Quantification of micropapillary component on transurethral resection is associated with likelihood of occult lymph node metastasis at radical cystectomy
Quantification of micropapillary component on transurethral resection is associated with likelihood of occult lymph node metastasis at radical cystectomy
 The use of nephron-sparing intervention does not appear to be compromised after a period of active surveillance for patients with cT1 renal masses
The use of nephron-sparing intervention does not appear to be compromised after a period of active surveillance for patients with cT1 renal masses
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


