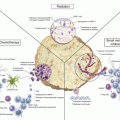
Fig. 8.1
DNA mismatch errors commonly occur at unstable genomic regions with multisatellite repeats. The repair of such DNA mismatch errors is the function of the MMR system. This process begins with recognition of DNA sequence errors by MutS homologues, which then recruits the MutL and associated peptides. The aberrant DNA bases are then excised and replaced by the replicative DNA polymerase. Abbreviations: Exo1 exonuclease, PCNA proliferation cell nuclear antigen
Much of our understanding of MMR function, and the pathology resulting from loss of function has been collated from preclinical models, which have indicated a degree of functional overlap between the mismatch repair components. MSH2 appears to be essential for the initial recognition of DNA errors: Msh2 −/− mice have a strong mutator phenotype at a cellular level, and develop lymphoma at an early age [23]. Furthermore, mice with conditional Msh2 loss in the intestine develop multiple intestinal tumours [50]. Msh6 null mice, similar to Msh2 −/− , are prone to lymphoma development, but do not exhibit the same predisposition to intestinal tumourigenesis; whilst Msh3 −/− mice are far less prone to cancer. However, combining Msh6 and Msh3 ablation resulted in intestinal tumour formation and a phenotype similar to Msh2 deficiency [24, 28]. These findings broadly correlate with observations in human cancers. In HNPCC families, Msh6 germline mutations are uncommon, and Msh3 −/− mutations are not seen. This suggests a degree of redundancy between MutSα and Mutβ. Simultaneous loss of both is required for a MMR-D state and cancer development [24]. Similar murine knockouts have also delineated the role of MutL proteins. Mice with Mlh1 deficiency develop multiple intestinal adenomas and adenocarcinomas, in addition to other epithelial cancers and lymphoma. Psm2 −/− mice, however, only showed predisposition to lymphoma and sarcoma development [79]. It is suggested that PSM2 loss can be compensated by the function of MutSβ, a heterodimer of Psm1 and Mlh1 [81].
While loss of MMR function may be evolutionarily beneficial for microbes by increasing genetic variation with subsequent selective advantage, as evidenced above, its consequences in mammals tend to be highly detrimental. Inherited mutations in mismatch repair genes results in Lynch syndrome, an autosomal dominant condition that confers a predilection to malignancies of the colon and rectum, endometrium, stomach, ovary, prostate and several other sites [60]. The lifetime risk of colorectal cancer for patients with Lynch syndrome is between 20 and 50 % depending on the inherited variant and sex of patient [26, 35, 80]. Several large studies have shown that it accounts for approximately 3 % of all CRCs in unselected series [37, 73]. Patients with Lynch syndrome inherit a mutation in single copy of one of the mismatch repair genes, most frequently MSH2 though any of the genes (MLH1, PMS1 or PMS2) may be mutated. During adult life a somatic cell within the colon loses the wild-type copy as a result of a “second hit” – comprising either loss of heterozygosity, somatic mutation or hypermethylation of the gene promoter region [44, 104]. In sporadic CRCs, MMR deficiency is almost always caused by hypermethylation of the MLH1 promoter with consequent silencing of gene expression [113].
Consistent with its essential role in the maintenance of genomic stability, loss of MMR function leads to an increased frequency of mutations across the genome (typically 10–100 mutations per Mb), referred to as “hypermutation”, and increased risk of carcinogenesis [7, 75]. Genomic regions at particular risk of mutation as a result of defects in mismatch repair include long repetitive mononucleotide or dinucleotide sequences, known as microsatellites [51]. These microsatellites are particularly susceptible to small insertion and deletion (indel) mutations as a result of polymerase slippage during DNA replication, and the MMR system is the predominant mechanism by which these errors are recognised and repaired. Failure to do this causes phenotype referred to as microsatellite instability (MSI) [58, 76]. Clinically, mismatch repair deficiency has typically been diagnosed by immunohistochemistry for mismatch repair protein expression, or by PCR of microsatellite markers to confirm instability [54, 73, 92, 109]. It has recently also been shown that MMR deficiency can be identified by quantification of mutational load using NGS cancer panels. In a study that used either 341 or 410 gene panel assay, a cut off values >20 mutations but <150 proved a 100 % sensitive and 100 % specific method for the detection of MMR-D [99].
Clinical Features of MMR-D Colorectal Cancers
In addition to their distinct biology and mutational profile compared to other tumours, MMR deficient CRCs also display distinctive clinical and pathological characteristics. They typically occur in the proximal colon [103], and are poorly differentiated [2]. Perhaps most relevant to this Chapter, MMR-D cancers are also associated with a substantially increased number of tumour infiltrating lymphocytes (TILs), and particularly CD8+ cytotoxic lymphocytes when compared to other CRCs [2, 25, 97, 117].
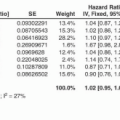
The association of mismatch repair deficiency with colorectal cancer prognosis has been the subject of much research. Though most early studies were underpowered to detect a difference in outcome, a landmark meta analysis published in 2004 by Popat and co-workers combined data from 32 studies to confirm that MMR-D was associated with a significantly reduced risk of cancer recurrence in stage II/III colorectal cancer, with a hazard ratio of 0.67 for disease-free survival [77]. Subsequent studies have both confirmed this result, and revealed that the impact of MMR-D on prognosis appears to vary according to the stage of disease. In stage II disease, MMR-D tumours have roughly half the risk of recurrence of other cancers, while in stage III disease the reduction is more modest (HR of ~0.8) [6, 43, 85, 96]. Interestingly, the 4 % of stage IV tumours with MMR-D have a dismal prognosis, with an outcome that is worse than that of any other molecular subtype in several studies [32, 36, 106].
It has been generally believed that the favourable prognosis of early stage MMR-D colorectal cancers is a direct consequence of the enhanced immune response they appear to elicit – a conclusion supported by recent studies which have demonstrated that the strength of the adaptive immune response correlates with decreased metastases and increased survival time [30, 70]. However, this begs the question as to how these tumours are able to grow into clinically detectable cancers when they appear to induce such a potent immunological reaction? A highly plausible answer to this apparent paradox was provided by an elegant recent study, which demonstrated that in addition in increased cytotoxic markers, MMR-D tumours also display substantial upregulation of immune checkpoints, such as PD-1, PD-L1 and CTLA-4, which counteracts the host cytotoxic immune response [56]. The possibility suggested by these data – that these cancers would be particularly good candidates for treatment with immune checkpoint inhibitors was explored in a prospective clinical trial conducted by the same group [55], the exciting results of which are discussed further subsequently.
POLE Proofreading Domain Mutation
In addition to the mismatch repair system, eukaryotes have evolved several other mechanisms that function to suppress potentially deleterious mutagenesis. One of these that has recently emerged as important in human cancers is the proofreading exonuclease activity intrinsic to the replicative DNA polymerases Pol δ and Pol ε, encoded by POLD1 and POLE in humans [82]. During DNA replication, proofreading of the newly synthesised strand serves to recognise and excise mispaired bases incorrectly incorporated by the polymerase catalytic domain, following which the correct base can be inserted, and replication continued [82]. While studies in yeast and in mice have confirmed that loss of either Pol δ and Pol ε proofreading activity results in a mutator phenotype and causes cancer [65, 95], evidence that such defects are relevant in human tumours has been limited. However, in 2012 both TCGA and another study demonstrated that a subset of highly mutated, but mismatch repair proficient colorectal cancers harboured recurrent mutations within the POLE exonuclease domain [89, 102]. In parallel with this report, a study of Individuals with intestinal polyposis and family history of colorectal cancer but without Lynch or other predisposition syndromes, demonstrated that these patients harboured germline mutations in the exonuclease domain of POLD1 or POLE [71].
Both the somatic and germline POLE exonuclease domain mutations affect highly conserved residues close to the DNA binding interface, and causality of several of these variants has been further supported by the demonstration that they confer a mutator phenotype in yeast (see Rayner et al. for a recent review). In addition to colorectal cancer, somatic POLE exonuclease domain mutations also occur in 6–15 % of endometrial cancers [14, 19, 20, 100] and rarely in tumours of the stomach, pancreas and brain where they are also associated with ultramutation, microsatellite stability and characteristic mutation spectrum [82, 93].
Recent studies have shown that endometrial cancers with ultramutation caused by POLE proofreading domain mutations have an excellent prognosis, despite a strong association with high-grade histology – a recognised poor prognostic factor. Interestingly, like MMR-D colorectal cancers, POLE-mutant endometrial tumours show evidence of a striking cytotoxic T cell response, and strong upregulation of immune checkpoints [110]. Given these data, the results of similar study of the consequences of POLE proofreading domain mutation in colorectal cancer are eagerly awaited.
Determinants of the Anti-tumour Immune Response
To date, most mechanistic data pertaining to the interaction between tumours and the host immune response, and the mechanism of action of immune checkpoint inhibition relates to the ability of host T cell compartment to recognise and target malignant cells [86, 87]. The postulate that such discrimination could occur as a consequence of recognition of tumour antigens as non-self was first suggested by seminal studies performed more than half a century ago. Sarcomas were induced in a mouse using methylcholanthrene, then subsequently excised and used to produce a tumour cell suspension. Inoculating syngeneic mice with this suspension resulted in rapidly growing tumours, which were then also removed. When these same “exposed” mice were re-inoculated with the same tumour mixture, there was evidence of resistance and in some cases complete tumour regression [34]. Subsequent work confirmed these original findings: showing that it is possible to induce anti-tumour immunity [78]. Importantly, it was shown that the acquired immunity was specific to the tumour cells, and not a consequence of genetic heterozygosity between the donor and recipient mice [21].
The T-cell is central to the adaptive immune response, and has numerous vital functions in both tumour recognition and elimination. T-cell maturation and development occurs in the thymus, where they begin life as double negative (CD4− CD8−) precursor thymocytes. Somatic rearrangement of the T-cell receptor (TCR) loci, first TCRβ and then TCRα, leads to the generation of T-cells with unique TCRs. Following TCRαβ heterodimer expression, thymocytes then undergo positive and negative selection. Those cells expressing Major Histocompatibility Complex (MHC) class II restricted receptors generally become CD4+ cells, while those expressing class I restricted are skewed towards CD8+ characterisation. Thymocytes that exhibit strong reactivity to self-antigens are clonally deleted, a process referred to as negative selection or central tolerance. The activation of T-cells occurs through interaction of the TCR with peptides presented by Major Histocompatibility Complexes (Major Histocompatability Complexes, MHC) of either class I or II that are present on cell surfaces. The specific part of a peptide that acts as an antigen and is capable of eliciting an immune response is referred to as an epitope. This process is aided by the co-receptor CD4 in T-helper cells, and CD8 in cytotoxic T-cells [33].
MHC class I molecules are expressed on the surface of most cells and are capable of being directly “read” by CD8+ cytotoxic T cell, a process which can result in direct killing of the offending cell. In contrast, MHC II is found on professional Antigen Presenting Cells (APCs) and serve to stimulate CD4+ T helper cells (Th). Under normal conditions, T cells do not react with self-peptides presented by HLA class I. However, foreign antigens presented by class I molecules may be recognised as non-self and induce a T cell response.
Current evidence suggests two principal mechanisms by which tumour peptides can act as antigens. The first category includes peptides that arise from aberrant expression of non-mutated self-proteins in cancer cells. The best described of these are referred to as cancer testis (CT) antigens, because their expression is restricted to the testis under normal conditions. The testis is an immunologically privileged site, and presentation of antigens at this site is associated with T cell tolerance due to expression of co-inhibitory molecules. Outside the testis, cancer testis antigens are usually not expressed, but aberrant expression occurs in several human cancers. Because these sites lack immune privilege T-cell tolerance is incomplete, and consequently expression of CT antigens by tumour cells represents a potential immune target. Much research has focused on vaccination against CT antigens commonly expressed in certain tumours, including MAGE1 in melanoma and New York oesophageal squamous cell carcinoma (NY-ESO-1) antigen in various tumours such as melanoma, bladder, prostate and hepatocellular [18, 45]. While the results of early studies were mixed, recently this approach has been associated with some success [17, 22, 45, 61]. However, there is little evidence that CT antigens play a substantial role in colorectal cancer, and they will not be further discussed in this Chapter. In contrast, most current and emerging evidence indicates a far more important contribution for the second class of tumour antigens, which include peptides encoded by DNA sequences that are normally absent from the human genome, a group collectively referred to as neoantigens.
Neoantigens in Cancer
There are two principal mechanisms by which novel antigens are generated in cancers. The first occurs in the substantial fraction of human cancers that are a consequence of viral infections. While the hijacking of normal cellular processes by cancer-causing viruses enables the cellular proliferation and survival that leads to malignancy, the resultant tumours often display epitopes derived from viral open reading frames integrated in the host DNA [48, 67, 72, 114]. Because these viral sequences are not normally found in the human genome they can be recognised as non-self, and targeted by T cells. Many chronic viruses have evolved mechanisms to suppress this immune response, including downregulation of MHC class I molecules on infected cells [4, 101], and immunosuppression through upregulation of PD-1 [5] and IL-10 [10]. However, recent data strongly suggest that an immune response against viral epitopes may account for the favourable prognosis of several viral-associated malignancies, including those in the upper GI tract such as Epstein Barr Virus (EBV)–associated gastric cancer [13]. However, like CT antigens, there is little evidence that viruses contribute to the pathogenesis of colorectal cancer, or that viral antigens contribute to its prognosis. The second mechanism relates purely to the novel peptides that arise from tumour-specific somatic DNA mutations, often referred to as tumour neoantigens. Because such neoantigens are completely absent from noncancerous tissues, T-cells reacting to them are not subject to the rigours of central and peripheral tolerance, and consequently are capable of eliciting a more vigorous immune response. Furthermore, their tumour-restricted expression means that therapies targeting these variants should be highly tumour-specific with lower rates of off target deleterious side effects. The last few years have witnessed substantial advances in the understanding of the determinants of immunogenicity of neo-antigens, and rapid progress in the ability to identify and predict these variants in cancers, largely as a consequence of improvements in sequencing technology and bioinformatics. These advances are reviewed in the subsequent sections (Fig. 8.2).
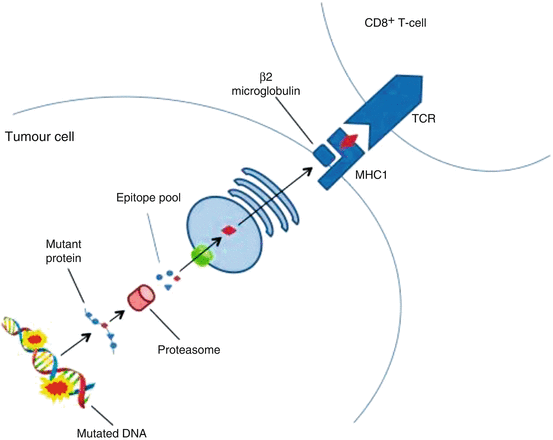
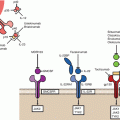
Fig. 8.2
Simplification of neoantigen presentation to CD8+ cells. Tumours harbour numerous non-synonymous, protein altering, DNA mutations that provide a portfolio of possible novel epitopes. These mutated peptides undergo intracellular processing, first being catabolised by proteasomes in the cytoplasm. In the Endoplasmic reticulum, these putative epitopes are bound to MHC class I molecules: the most selective step in determining the antigenicity of a mutant peptide. This protein complex is then translocated to the cell surface where it is presented to the T-cell receptor of a CD8+ cytotoxic T-cell
Identification of Tumour Neoantigens
The ability to predict which tumour mutations are likely to generate antigenic epitopes results from a combination of advances in understanding the mechanisms of peptide presentation by MHC molecules, the advent of next generation sequencing, and bioinformatic tools and pipelines to combine both for individual tumours.
Peptide binding with HLA-1 appears to be the most discerning step in the antigen presentation process. Each individual MHC molecule presents a unique binding specificity to a given peptide. The genomic region encoding for MHC is highly polymorphic, and to date more than 3000 allelic variants have been discovered. Of the 1500 known HLA class 1 molecules, less than 5 % have been examined in relation to their binding with more than 50 peptides [59, 90]. Because experimentally characterisation of the binding specificities of peptides for HLA molecules is laborious, the availability of a data driven bioinformatic approach would be of great benefit. NetMHCPan, developed by Nielsen and colleagues, is one such tool [69]. Using peptide and primary HLA sequencing as inputs, this artificial neural network (ANN) is able to predict the affinity of any peptide for HLA-A or HLA-B molecules. The authors validated this in silico approach by predicting the affinity of 500,000 random peptides for both HLA molecules. Of these, they selected 10–15 with high affinity (>50nM) and confirmed the interaction using an in vitro assay. NetMHCPan was able to predict binding with 86 % accuracy. Further validation was carried out on peptide binding with previously unstudied HLA molecules, and with known HIV immune epitopes. This approach has also been validated for use in non-human primates [42].
As discussed previously, next generation sequencing technology provides unprecedented ability to examine the mutational landscape of cancers. In particular, study of tumour exomes, that is the 1–2 % of the genome that codes for protein, is now both relatively inexpensive and scalable, albeit in specialist centres. With this wealth of available data, the next step has been to understand how the tumour mutational landscape defines the neoantigen repertoire. Although a typical tumour genome may contain 10–100 s of SNV or indel mutations that result in amino acid substitutions, only a small percentage of these result in the expression of neoantigens [39]. To identify these, most investigators have used an in silico approach informed by the understanding of MHC I binding preferences as described.
In an early study that predated the development of NGS approaches, Segal and colleagues sought to predict the neoantigen repertoire of 11 CRC and 11 breast cancer samples, from somatic mutations in roughly 13,000 cancer genes. Focusing solely on theoretical binding of tumour-derived 9mer peptides with HLA-A*0201 (the most common class I allele in Caucasian populations), they found that CRC and breast cancer possessed an average of seven and ten predicted antigenic novel epitopes respectively; corresponding to approximately 1 antigenic epitope for every ten nonsynonymous mutations [88]. As humans have up to six different MHC I alleles (2 loci for each of HLA-A, B and C) the authors suggested that their conservative estimate could be multiplied sixfold, resulting in a possible average of 40–60 novel epitopes in typical colorectal cancers. While this study provided an intriguing insight into the possible antigenicity of mutations in cancer genes in these common tumour types, it was necessarily limited by the technologies of the time. For example, the absence of gene expression data made it uncertain as to whether all these mutant peptides were in fact expressed, and the absence of individual HLA typing meant that the binding affinities could only be calculated against a single representative class I molecule.
Building on these results, Brown and colleagues developed a bioinformatic tool, HLAminer to extract permit four digit HLA typing from TCGA RNAseq data. They combined these data with exome sequencing from patients with six common tumour types (lung, ovary, breast, brain, colorectal and kidney) analysed by TCGA. For each missense mutation, they calculated the binding affinity for the patients HLA class I molecules using NetMHCPan. Informed by the results of the preclinical studies discussed earlier, they defined antigenic mutations as those that gave rise to peptides that bound with high affinity (<500nM). This approach suggested that an appreciable fraction of missense mutations were antigenic, and revealed substantial variation in the number of antigenic variants both between and within different tumour types. Importantly, the number of immunogenic mutations correlated significantly with CD8+ cytotoxic response and survival rates, and interestingly, the strongest association was observed among the 170 cancers included in this study [11]. An extremely comprehensive subsequent study by Rooney and colleagues also used TCGA data to interrogate the burden and correlates of antigenic mutations in multiple cancer types. To perform their analysis, the authors developed a novel bioinformatic tool, Polysolver, which permits HLA typing with four digit accuracy from exome sequencing [84]. In a separate publication [94], the group demonstrated that Polysolver was able to call HLA type with substantially greater accuracy than HLAminer and comparable to conventional serological and PCR typing strategies [94]. The group subsequently employed Polysolver to type the class I HLA alleles in over 7000 cases comprising 18 different types of tumour from TCGA, and in parallel used exome sequencing data to calculate all possible peptides predicted to be generated by all missense and indel mutations in these patients’ tumours. Restricting their analysis to variants in expressed genes they demonstrated that on average 50 % of non-silent mutations were predicted to generate an predicted neoepitope, defined as a peptide predicted to bind the patient’s class I alleles with affinity of <500nM. The number of antigenic mutations displayed a strong positive correlation with the level of tumour immune cytolytic activity, as quantified by the geometric mean of the cytotoxic effectors granzyme A (GZMA) and perforin (PRF). After noting that the number of antigenic mutations in tumours correlated very strongly with their total mutation burden (Spearman rho = 0.91), the authors were able to demonstrate that in both renal and colorectal cancers, the observed number of predicted neoepitopes was significantly lower than predicted based on the frequency of silent mutations in these cancers, suggesting immune selection against antigenic variants. Importantly, this depletion was lost when neoepitopes were estimated based on randomly shuffled, rather than individual HLA genotypes [84].
Functional Validation of Predicted Neoepitopes
While the power of in silico approaches to predict antigenicity has been demonstrated by the studies discussed above, it is important to note that only a small fraction of mutations predicted to be antigenic by bioinformatic pipelines are confirmed to be so when analysed in functional studies [87]. Castle and colleagues performed next generation sequencing of B16F10 cells, a mouse melanoma cell line that is able to grow when inoculated into immunocompetent syngeneic hosts, and identified 962 mutations in 563 expressed genes. 50 of these variants were selected for functional study, based on expression and predicted antigenicity, although this approach did not include the predicted binding of the peptides for MHC class I molecules. Using a long peptide vaccination strategy in mice, 16 of these variants proved to induce an adaptive immune response when inoculated in vivo. Importantly, vaccination with several of these peptides protected against tumour growth following subsequent inoculation of B16F10 cells [16].
One such strategy to improve the accuracy of neoantigen prediction is to combine the sequencing and bioinformatic predictions with the use of mass spectrometry analysis. Examining the mouse MC38 colorectal cancer cell line, Yadav and colleagues used whole exome sequencing to identify 4285 non-synonymous mutations, 1290 of which were in expressed genes. 170 were considered candidate neo-epitopes in that they were predicted to bind MHC class I molecules using NET MHCPan [116]. In parallel with the informatics approach, the authors employed an additional strategy to confirm the immunogenicity of candidate immunogenic peptides. Using mass spectrometry, they discovered that only of the seven mutated peptides were found to be presented on MHC I. These seven epitopes were then subjected to further assessment using computational structural modelling to assess MHC binding affinity, and interaction between TCR and the mutated peptides. These experiments suggested that of the seven peptides, only three would meet the criteria to stimulate a T-cell response. Indeed, inoculation of mice with these three selected peptides induced a CD8+ T-cell response, conferring immunity to tumour grafts and an immune mediated growth control of already established tumours [116].
While most studies have focused on the role of cytotoxic T cells in recognition of neoantigens, emerging evidence suggests that T helper cells also serve an important role in both this and the antitumour immune response more broadly. Activated via MHC II, CD4+ Th cells participate in augmenting CD8+ response and recruiting natural killer (NK) cells. Using a similar approach to selecting MHC I class presented mutations, melanoma intra-tumour CD4+ cells could be strongly induced in response to autologous neo-epitopes [57]. Notably, a recent report suggests that MHC II class restricted epitopes may be far more abundant in tumours than MHC I class I restricted peptides. In analysis of melanoma, mammary and colorectal murine cancer models Kreiter and colleagues demonstrated that more than 80 % of nonsynonymous mutations were MHC II class restricted. These conclusions were corroborated by analysis of tumours from TCGA samples. The relative abundance of CD4+ responsive mutations may be due to less stringent length and sequence criteria for MHC II binding when compared to MHC class I restricted peptides. Additionally, incorporating these MHC II mutant epitopes into a poly epitope RNA based vaccine generated efficient tumour control in mice, inducing a combined CD4+ and CD8+ immune response [49]. It would appear that there exists a more extensive range of MHC II epitopes; specific CD4+ cell targeting of these mutations is central to an effective anti-tumour immune response [108]. Figure 8.3 summaries the experimental process to identifying and validating potential tumour associated neoantigens.
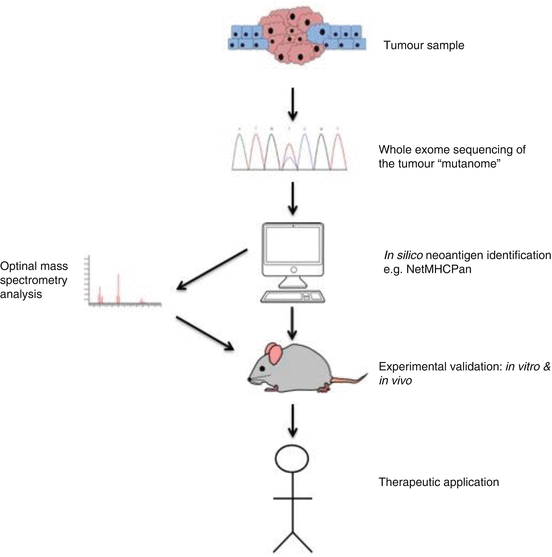
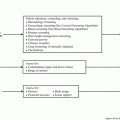
Fig. 8.3
Tumour associated neoantigen identification pipeline. Representative tumour samples are subjected to whole exome sequencing to identify possible neoantigen forming DNA mutations. In silico analysis selects candidate neoepitopes based on the predicted binding with HLA molecules. The use of additional algorithms such as HLAminer or Polysolver to define the patient specific HLA allele further refines this process of selection. Additionally, mass spectrometry adds another possible filtering step. The candidate neoantigens are then experimentally validated before possible therapeutic application, which includes vaccination or autologous T-cell transplantation
Neo-Epitopes in Colorectal Cancers
Given the correlation between the total mutation burden of cancers and the number of predicted neoepitopes they are predicted to generate, it is unsurprising that most attention has focused on the subset of immunogenic, hypermutated, mismatch repair deficient tumours.
In a recent study, Mlecnik and colleagues demonstrated that increased intratumoural immune gene expression in mismatch repair deficient colorectal cancers was associated with a significantly greater number of predicted neo-epitopes in this molecular subset. Interestingly, similar to the previous study by Rooney and co-workers, they also noted that the number of neo-epitopes in mismatch repair deficient tumours was less than predicted by the frequency of silent mutations, suggesting that antigenic variants were subject to negative selection in vivo [64]. This analysis also suggested that the enhanced immunogenicity of mismatch repair deficient tumours may also result at least partly from their enrichment of frameshift mutations, as these variants were predicted to generate more neoepitopes per mutation than missense variants.
In general, the prediction that mismatch repair deficient cancers are to likely generate substantially more neo-epitopes than mismatch repair proficient tumours is mirrored by their apparently enhanced immunogenicity. However, notable exceptions exist – that is some mismatch repair deficient tumours are predicted to generate many neo-epitopes but fail to demonstrate significant immune response, while a subset of mismatch repair proficient cancers appear strongly immunogenic despite seeming to harbour a paucity of neoepitopes. Indeed, in an elegant recent study from the Rosenberg group, exome sequencing was performed on metastases from patients with several different tumour types including colorectal cancer. They proceeded to examine whether somatic mutations from tumours were recognised by tumour infiltrating lymphocytes using a tandem minigene approach, and confirmed reactivity of several mutations, including a KRAS G12D mutation in a patient with colorectal cancer. Importantly, none of the colorectal cancers examined in this study were hypermutated (the number of mutations per colorectal cancer sample ranged from 58 to 134) [107].
While this study clearly demonstrates that non-hypermutated colorectal cancers may harbour reactive neoepitopes, the paucity of responses to anti-PD1 therapy in unselected colorectal cancers (one of 33 patients in trials conducted by Topalian and colleagues [8, 105]) contrasts with the dramatic responses among mismatch repair deficient tumours [55]. Whether this discordance is a reflection of the lower probability that a non-hypermutated tumour harbours a highly antigenic variant, or the cumulative effect of multiple modestly antigenic neoepitopes in hypermutated cancers awaits definition.
Immunoediting of Neoepitopes
Until recently, the interaction between the host immune system and malignancy was often referred to as immunosurveillance – a term that implied that the immune response had a purely protective role in preventing cancer development. However, during the last few years it has been broadly accepted his term fails to capture what is clearly a complex, dynamic interaction between the two [86]. The seminal studies of MCA-induced murine sarcomas discussed earlier demonstrated that the immune response influences not only the kinetics of tumour growth, but also the neoantigen landscape of the developing malignancy. In an elegant series of experiments, the investigators demonstrated that sarcomas grown in Rag2 −/− immunodeficient mice were frequently rejected when transplanted into immunocompetent wild-type mice, while those transplanted from either wild-type or Rag2 −/− into Rag2 −/− recipients grew progressively. Furthermore, in this study the authors were also able to demonstrate that tumour rejection was a T-lymphocyte dependent process, and to identify the specific rejection antigens by exome sequencing. Taken together, these results provide convincing evidence of the protective role of lymphocytes against tumour development, and the ability of the host immune response to sculpt the mutational landscape of the developing tumour – a process they referred to as “immunoediting” [91].
The process of immunoediting has been hypothesised to involve three key phases: elimination, equilibrium and escape. Elimination is the process by which the immune system is able to detect and remove nascent cancer cells. While direct in vivo evidence to support this stage is lacking, it has been inferred from numerous murine models in which facets of the adaptive or innate immune system – such as lymphocytes, or immune effector molecules – have been selectively ablated (Reviewed in: [111]). Tumour cells that escape initial elimination may then enter a steady state of equilibrium, during which the immune system prevents tumour progression, resulting in dynamic modelling of the tumour immune landscape. It has been speculated that tumours may exist in this state for some time before becoming clinically evident. Overt disease eventually results when tumour cells are able to circumvent the controlling effects of the immune system: escape. The final product, the cancer and its neoantigen profile, is the result of the accumulation of mutations and continuous immune pressures: this Darwinian selection produces clones that are “invisible” to the immune system. It has in fact been proposed that this final stage – the evasion of immunosurveillance – is the seventh hallmark of cancer [38, 120]. Current evidence suggests two principal methods by which this is achieved: by reducing the capacity for immune cell recognition, or alternatively through the creation of an immunosuppressive tumour microenvironment (TME). The former is often achieved by mutation or decreased expression of components in the antigen presentation machinery, including β2 microglobulin or the HLA genes, while the latter appears most commonly due to upregulation of immune checkpoints or other immunosuppressive molecules such as PTGS2 [63, 118, 119]. The process of immunoediting is shown in Fig. 8.4.
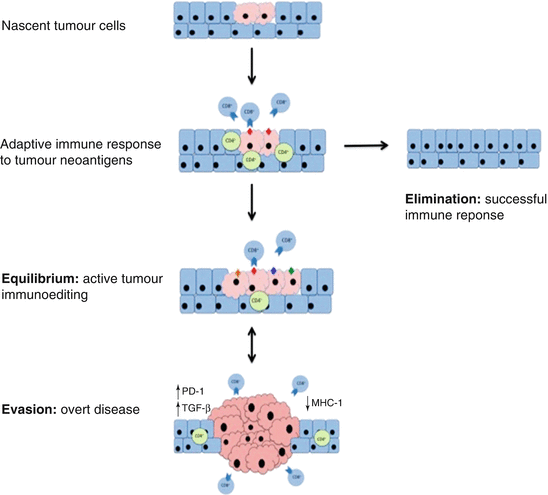
Fig. 8.4
The process of immunoediting. Nascent tumour cells display neoantigens that are recognised by the adaptive immune response, and results in CD4+ and CD8+ T-cells response (other facets of the immune system also participate but are not shown here). This immune response results in two possible outcomes: elimination, in which the tumour cells are completely eradicated; or alternatively a state of equilibrium develops, during which constant immune pressure serves to sculpt the neoantigen landscape by selecting against immunogenic clones and promoting the development of tumour cells invisible to the immune system. This process may continue for an extended time, and the tumour becomes only becomes clinically relevant when immune evasion occurs. In this third, and final, stage the highly edited non-immunogenic tumour is able to escape its immunological restraints: leading to invasion and dissemination
As discussed before, the depletion of neoepitopes in hypermutated mismatch repair deficient colorectal cancers provides convincing, if correlative, evidence of immunoediting of these tumours [64, 84]. Furthermore, both mutation of B2M and downregulation of HLA expression appear to be common events in mismatch repair deficient cancers, and a recent study has shown that these tumours are also characterised by robust upregulation of immune checkpoints including PD1 and PDL1 [56].
Targeting Neoantigens for Therapy
The enrichment of neoepitopes among hypermutated colorectal cancers is of substantial clinical relevance and is currently the subject of considerable investigation. As noted previously, because these variants are tumour-specific and absent from healthy tissues targeting them may provide a highly favourable therapeutic index and avoid the deleterious side effects of unselective traditional cytotoxic chemotherapeutics. Although most attention has focused on the use of immune checkpoint inhibitors, other therapeutic opportunities include the development of a tumour specific vaccines, and Adoptive Cell Therapy (ACT).
The striking benefit of PD1 blockade in patients with mismatch repair deficient colorectal cancers has been mentioned previously. In this landmark study, profound and seemingly durable responses were observed in seven of nine (78 %) patients with mismatch repair deficient tumours, compared with two of 18 (11 %) mismatch repair proficient cancers [55]. As expected, exome sequencing demonstrated that the number of somatic mutations in mismatch repair deficient cancers substantially exceeded that in mismatch repair proficient tumours (mean of 1782 mutations vs. 73 mutations; P < 0.001), as did the number or likely neoantigens, predicted using the patients’ individual HLA haplotype (mean 578 vs. 21 neoepitopes). Importantly, despite the small size of this study, high numbers of somatic mutations and tumour neoepitopes were significantly associated with longer progression-free survival following immune checkpoint blockade, and also demonstrated a trend towards improved response rate [55].
Related posts:

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


