Fig. 6.1
Schematic overview of the chapter discussion pertaining to the role of estrogen receptor (ER) α action in the maintenance of glucose homeostasis and insulin action for the protection against obesity and chronic diseases associated with metabolic dysfunction including atherosclerosis, type 2 diabetes, and certain forms of cancer
Molecular Mechanisms of Estrogen Receptor (ER) Action
Early studies in reproductive tissues investigating the actions of estradiol led to the paradigm of classical nuclear ERs as ligand-activated transcription factors (Fig. 6.2) [13]. Although ERs exist in two main forms, α and β, which have multiple splice variants of unknown function, ERs exhibit tissue specificity in expression and function [14]. The classical, or genomic mechanism of ER action, includes the ligand-activated dissociation of ER from its chaperone, receptor dimerization, and receptor binding either to estrogen response elements (ERE) in target genes promoters or to AP-1 or SP-1 response elements through association/tethering with other transcription factors bound to DNA (Fig. 6.2) [15]. After binding, ER dimers interact with basal transcription factors leading to activation or repression of target gene expression. Overlap in binding sites for E2-liganded ERα and ERβ is observed when receptors are expressed individually; however, when both ERs are present, few sites are shared. Each ER restricts the binding site occupancy of the other, with ERα dominating [16]. Moreover, ligand-activated ER promotes transcription in a cyclic fashion. The repeated cycling of the receptor complex on and off target promoters in the presence of continuous E2 stimulation may represent a mechanism of continuous sensing and adaptation to the external hormonal milieu to yield the appropriate transcriptional response [17].
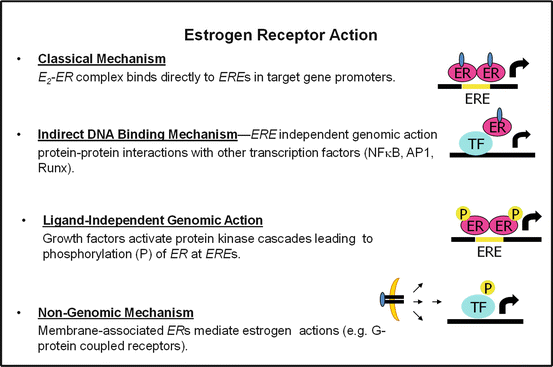
Fig. 6.2
Molecular actions of ERα to activate or repress target genes by classical, DNA binding or nongenomic actions. ERE estrogen response element in target gene promoters, P phosphorylation, TF transcription factor
In addition to classical signaling, E2-ERα can act within minutes or seconds via extranuclear and membrane-associated forms of the receptor (Fig. 6.2) [18]. Membrane associated receptors are localized to caveolae where they congregate with other signaling molecules, including G proteins, growth factor receptors, tyrosine kinases (Src), linker proteins (MNAR), and orphan G-protein coupled receptors (GPCRs). In a variety of cell types, membrane and extranuclear pools of ERs activate protein kinases that phosphorylate transcription factors to promote their nuclear translocation and transcriptional action [18, 19]. The G protein-coupled estrogen receptor (GPER), or GPR30, has been reported to respond to E2 however its role as an ER is still controversial and thus will not be discussed in this chapter. Although it is thought that estrogen effects on reproductive function are almost exclusively mediated via classical nuclear ERs acting as ligand-activated transcription factors, a large component of ER action related to energy metabolism also involve extranuclear ERs (Fig. 6.2) [20]. A central question in the field pertaining to the tissue-specific sites of ERα action and the molecular mechanisms by which the receptor selectively activates or represses target genes persists.
The Impact of Brain Estrogen Action in the Regulation of Energy Intake and Expenditure
Studies in humans and animal models have established important roles for estrogens in the regulation of metabolism. As estrogen levels decline during and following menopause, the prevalence of obesity escalates markedly [11, 21, 22]. Ovariectomy (OVX), i.e. removal of the ovaries, leads to increased adiposity in rodents [23–25], that is prevented by estrogenization typically by subcutaneous implantation of a time-release estrogen pellet [26]. Although OVX induces a transient increase in food intake, prevented by E2 normalization [27, 28], hyperphagia cannot solely account for the changes in metabolism and the development of obesity [25]. Furthermore, mice of both sexes lacking the enzyme CYP19, aromatase (aromatase promotes the synthesis of estradiol), develop obesity in the absence of hyperphagia. Instead, aromatase-deficient mice exhibit reduced physical activity and diminished lean body mass [29]. Similarly, mice of both genders with a homozygous null mutation for Esr1 (ERα) develop obesity in the absence of hyperphagia [30, 31]. This work suggests that although endogenous E2 favors body weight homeostasis by increasing energy expenditure [32], exogenous estrogens may promote energy balance by influencing both energy intake and expenditure. Thus, loss of ERα action produces a predominant decrease in energy expenditure, while conversely, increasing ERα signaling by raising serum E2 concentrations both suppresses energy intake and increases energy expenditure as illustrated in Fig. 6.3 and discussed below.
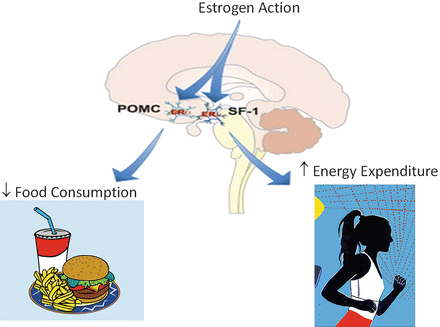
Fig. 6.3
The effects of ERα action in POMC and SF-1 neurons to control feeding and energy expenditure. Findings from Xu et al. Cell Metabolism 14, 453–465, 2011
For over a century we have known that specific regions of the CNS control food intake, energy expenditure, and weight gain, as lesioning of specific hypothalamic nuclei within the ventral medial hypothalamus (VMH) [33, 34] or lateral hypothalamus (LH) caused dramatic changes in these biological processes [35, 36]. ERα is highly expressed in the rodent brain including the ventrolateral portion of the VMN, the ARC, the medial preoptic area (MPOA), and the paraventricular nuclei (PVN) [37–43]. ERβ is expressed in the same hypothalamic nuclei but at significantly lower levels by comparison. The effects of E2 on energy balance are thought to be primarily mediated by ERα, as women or female mice with mutations in the ERα gene become obese [30, 44]. Moreover, ERα homozygosity in animals prevents the anti-obesity effects of estrogen replacement [26]. These gene deletion studies are consistent with pharmacological interventions in ovariectomized (OVX) mice in which the selective ERα agonist PPT was shown to suppress food intake, in contrast to the selective ERβ agonist DPN which failed to influence feeding behavior [45–47].
The signaling mechanisms of ER action in hypothalamic neurons are not fully defined, but evidence suggests the involvement of both classical and extranuclear ER actions [27, 48]. In the arcuate nucleus, ERα is highly expressed in pro-opiomelanocortin (POMC) neurons shown to modulate food intake, energy expenditure, and reproduction (Fig. 6.3) [49]. POMC neurons secrete α melanocyte stimulating hormone (αMSH), which acts in the PVN and lateral hypothalamus on melanocortin 3 and melanocortin 4 (MC3/MC4) receptors to produce catabolism by reducing food intake and increasing energy expenditure [50–53]. ARC POMC mRNA levels fluctuate over the course of the estrous cycle, with a marked increase occurring coincident with proestrous when E2 concentration is the highest [27, 54–57]. Conversely, POMC levels are reduced in ERα knockout mice [58]. These E2-induced synaptological rearrangements in POMC neurons are tightly paralleled with the effects of estrogens on food intake, energy expenditure, and body weight [27], and appear mediated by MC4R [59]. Indeed, deletion of ERα in POMC neurons of mice leads to hyperphagia without affect to energy expenditure or adipose tissue distribution [49].
The ventral medial hypothalamus, VMH, is also thought to be an important neural circuit responsible for the homeostatic regulation of body weight and food intake, as estrogens have been shown to directly alter the electrophysiological properties of VMH neurons [60]. Small hairpin (sh) RNA-mediated ERα gene silencing and selective ablation of ERα from SF-1 containing neurons of the VMH blunts E2-induced weight loss, promoting increased visceral fat deposition, and reductions in energy expenditure in the absence of hyperphagia (Fig. 6.3) [61]. These findings support the notion that ERα signaling in VMH neurons plays an important role in regulating physical activity, thermogenesis, and fat distribution.
Since leptin was first described in 1994 [62], it has proven to be a powerful catabolic signal in the brain, inhibiting food intake and increasing energy expenditure [52, 63, 64]. Leprb is localized in several brain areas including the VMH and the ARC, and colocalizes with several neuropeptides known to control food intake and reproduction [65–67]. Leprb also binds ERα in the ARC [68], which is consistent with estrogens inducing leprb mRNA [69], possibly by direct genomic action mediated by ERE binding in the promoter of the leptin receptor gene [70]. The extensive hypothalamic colocalization of these two receptors suggests a closely coupled interaction between peripheral derived signals in the regulation of energy homeostasis. Indeed, increased E2 is associated with enhanced central leptin sensitivity in rodents [71–73]. Although circulating leptin levels do not change appreciably during the estrous cycle, ARC leprb expression is highest during estrous and metestrous and is inversely correlated with neuropeptide Y (NPY) mRNA expression [69]. Moreover, OVX lowers the sensitivity to central leptin when compared to normally cycling females, and this leptin resistance is prevented by E2 replacement [73]. Analogously in male rodents, exogenous E2 administration also increases sensitivity to central leptin [73]. The differences in central leptin sensitivity caused by the presence or absence of estrogens may occur downstream of leprb transcription and translation, possibly at the level of STAT3, as E2 decreases food intake and increases energy expenditure, resulting in a reduction in body weight in leptin-deficient (ob/ob) and leptin resistant (db/db) mice of both sexes [27].
NPY is an effective anabolic peptide considering that central administration of NPY potently increases food intake and decreases energy expenditure and fat oxidation [74–77]. ARC neurons coexpress NPY mRNA and leprb protein. Leptin administration decreases, while leptin deficiency or leptin resistance increases NPY (and AgRP) mRNA, demonstrating that leptin is a critical determinant of ARC NPY function [78]. NPY neurons in the hypothalamus not only affect feeding, but also influence reproduction, therefore, E2 can modulate both of these neuroendocrine systems by regulating NPY gene expression, NPY Y1 receptor expression [79], and NPY release [80]. Additionally, NPY/AgRP neurons are required to mediate the anorexigenic effects of E2 and hypothalamic expression of NPY and AgRP is tightly regulated across the estrus cycle, with the lowest levels during estrus concomitant with the E2 peak and feeding nadir in wild-type mice [81]. Importantly, the cyclic changes in food intake and E2-induced anorexia are blunted in mice with degenerated NPY/AgRP neurons [81], indicating that neurons coexpressing NPY and AgRP are functionally required for the cyclic changes in feeding across estrous cycle and that NPY/AgRP neurons are essential mediators of the anorexigenic function of E2. Interestingly, it was shown that ERα is completely excluded from NPY/AgRP neurons in the mouse hypothalamus [81], suggesting that E2 may regulate these neurons indirectly via unknown presynaptic neurons expressing ERα.
ERα Expression in the Control of Adipose Tissue Distribution and Obesity Susceptibility
There is a well-described sexual dimorphism in body fat distribution as men have less total body fat but increased intra-abdominal adipose tissue compared with women who have more total fat but a higher proportion of gluteal/femoral subcutaneous (SC) adipose tissue which is shown to be less pathogenic [82–86]. Following menopause and the decline in circulating E2, women gain intra-abdominal fat and develop male-pattern adiposity ameliorated by estrogen replacement therapy [87–89]. Additionally, adipose tissue redistribution is reported in male–female transsexuals receiving estradiol supplementation in which increased SC fat relative to intra-abdominal adipose tissue is observed [90]. Thus, estrogens are thought to regulate fat distribution [90, 91], and this may contribute to improved metabolic profile in women receiving HRT.
Accumulation of central intra-abdominal or visceral fat, (“android,” or male-pattern obesity) is correlated with increased risk and mortality from diabetes and atherosclerosis [92]. Intra-abdominal adipose tissue is thought to be metabolically and functionally different from SC adipose tissue. Indeed, compared the SC fat, intra-abdominal adipose tissue has more efferent sympathetic axons and capillaries, per unit volume, and these capillaries drain into the hepatic portal system [92]. Surgical removal of intra-abdominal adipose tissue in humans results in decreased insulin and glucose levels [93] and prevents the onset of insulin resistance and glucose intolerance in male rodents [94]. In contrast, surgical removal of SC fat tissue of equal weight has no appreciable impact on the same parameters [94]. Teleologically, males may preferentially deposit adipose tissue in the intra-abdominal depot because of its ability to be rapidly mobilized providing a quick and abundant energy source during times of increased energy demand. In contrast, SC adipose tissue allows for efficient storage of maximal calories per unit volume of tissue. Lipid deposition into SC adipose tissue may provide an evolutionary advantage for females by extending protection during limited caloric supply in order to maintain reproductive function. Importantly, females also mobilize adipose tissue stored in SC depots to meet the caloric demands placed on the body during breast feeding. In contrast to android or male-pattern adiposity, “gynoid” or female-pattern fat deposition is only weakly correlated with metabolic dysfunction and disease risk [95–99]. In fact, transplantation of SC fat from donor mice into visceral regions of recipient mice decreases total fat and improves glucose homeostasis suggesting that adipose-secreted factors may act systemically to improve metabolism [100].
In addition to ovarian production, estrogens are also synthesized in adipocytes (via aromatization of androgenic precursors by CYP19), and circulating levels are elevated in proportion to total body adiposity [101, 102]. ERα is highly expressed in adipose tissue [103, 104], and targeted global deletion of the ERα gene (ERαKO) in mice of both genders promotes increased adiposity, with a near doubling of the visceral depots compared with age-matched wild-type (WT) mice (Fig. 6.4) [30]. In contrast, mice with a homozygous deletion of ERβ (βERKO) show a body composition identical to WT mice, suggesting that ERβ may play a limited role in adipose tissue development and metabolism [105]. Reduced ERα expression or impaired ERα function due to genetic alteration has been linked with increased prevalence of specific features of the metabolic syndrome including obesity in both male and female humans and rodents [106–113]. The role of ERα in adipocytes and the phenotypic outcomes of adipose-specific ERα deletion in mice are currently under investigation by several laboratories around the world. Whether the obesity phenotype observed in whole body Esr1−/− mice or women harboring an ESR1 inactivating mutation is explained by loss of ERα from adipose tissue specifically or as a result of a secondary phenotype of ERα deletion from other metabolic tissues remains unknown.

Fig. 6.4
Metabolic dysfunction in ERαKO mice. Animals develop insulin resistance, impaired glucose tolerance and obesity as early as 3–6 months of age and are more susceptible to the deleterious effects of HFD than age-matched WT counterparts
Estrogen Action and Insulin Sensitivity
Insulin resistance is a central disorder in the pathogenesis of type 2 diabetes and a defining feature of the Metabolic Syndrome, a clustering of metabolic abnormalities including obesity, hypertension, glucose intolerance, and dyslipidemia [114, 115]. Normally cycling women show enhanced insulin sensitivity compared to men when normalized to lean mass, and this is a likely contributor to the reduced incidence of type 2 diabetes in pre-menopausal women [116, 117]. Moreover, although a 40–50 % reduction in insulin-mediated glucose disposal is consistently observed in male mice following high fat feeding [118, 119], E2-replete females, humans and rodents, are typically protected against a high fat diet and acute fatty acid-induced insulin resistance [120–123]. Following menopause or OVX, a precipitous decline in insulin sensitivity coincides with increased fat mass, and elevated circulating inflammatory markers, LDL, triglycerides, and fatty acids [11, 124, 125]. OVX mice and rats are insulin resistant, show impaired exercise-stimulated glucose disposal into muscle [126], and are more susceptible to the deleterious effects of high fat diet or lipid oversupply, and these physiological consequences of OVX are prevented by restoration of circulating estradiol within a physiological concentration [127].
Although chronic administration of E2 is shown to improve insulin sensitivity, at least in rodents, the acute action of E2 to promote insulin-stimulated glucose uptake into muscle remains disputed; this despite consistent observations of E2-induced activation of Akt and AMP-activated Protein Kinase (AMPK) [128, 129]. Furthermore, although administration of intravenous conjugated estrogens and E2 to postmenopausal women or OVX rats, respectively, is shown to stimulate a significant increase in glucose disposal during hyperinsulinemic-euglycemic clamp studies [130, 131], ex vivo treatment of skeletal muscle with E2 failed to recapitulate the same increase in insulin-stimulated glucose disposal [128]. This is also in contrast to short-term estradiol effects on insulin action in myotubes from postmenopausal women and age-matched men [132]. Thus, two questions remain; does E2 enhance insulin sensitivity and what are the critical tissues of E2 action that confer protection against insulin resistance induced by nutrient oversupply?
Similar to findings for ovarian failure in women and rodents, a reduction in circulating estrogens resulting from rare inactivating mutations or experimental deletion of Cyp19 (aromatase) in mice confers an obesity-insulin resistance phenotype [29, 106, 133–139]. The physiological and genetic evidence argues that E2 and ER favor insulin sensitivity in rodents and humans of both sexes when E2 is maintained within a tight physiological concentration. Indeed, replacement or augmentation of E2 to supraphysiological levels or over-stimulation of ER is thought to induce insulin resistance secondary to hyperinsulinemia and or a reduction in total GLUT4 expression in muscle [140, 141]. In fact, two studies have reported that in postmenopausal women, higher plasma levels of E2 were prospectively associated with increased risk of developing T2D [142, 143]. Clearly, additional studies in rodents and humans using a dose response strategy are necessary to better understand the interplay of steroid hormones including E2, testosterone, and progesterone on the regulation of metabolism and insulin action in glucoregulatory tissues.
Although, several laboratories have characterized the whole body ERαKO mouse (Fig. 6.4 Phenotype Overview), many questions still remain including the tissues responsible for conferring the severe insulin resistance-obesity phenotype. Does obesity arise from loss of ERα within adipocytes specifically or can it be driven as a secondary phenotype resulting from a loss of ERα in brain, skeletal muscle, liver, or even selective immune cells? Furthermore, does a loss of ERα specifically from myocytes drive skeletal muscle insulin resistance, or does this phenotype arise as a secondary consequence of increased adiposity and altered adipokine/cytokine release?
Although two forms of the receptor are expressed in many of the glucoregulatory tissues, ERα is found in much higher abundance than ERβ, as ERβ transcript is nearly undetectable in human and rodent muscle [132, 144, 145]. Consistent with these observations, homozygous deletion of ERβ failed to produce insulin resistance [44] in contrast to the marked skeletal muscle insulin resistance observed in ERαKO animals (Fig. 6.4) [146, 147]. The underlying mechanism contributing to impaired insulin action in ERαKO animals remains disputed as findings reported by Bryzgalova et al. [148] suggesting reduced total GLUT4 levels in muscle as an underlying cause for the ERαKO insulin resistance phenotype was not supported by Ribas et al. [146]. Furthermore, despite maintenance of GLUT4 mRNA and protein, Ribas et al. reported more dramatic skeletal muscle insulin resistance in ERαKO mice than Bryzgalova et al. work by Hevener and colleagues suggests that the skeletal muscle insulin resistance observed in ERαKO mice is predominantly due to direct effects of ERα deletion on pro-inflammatory signaling and proximal insulin signal transduction.
Indeed, emerging findings in muscle from muscle-specific ERα knockout mice and myotubes with ERα knockdown show no alteration in GLUT4 mRNA or protein despite a marked impairment in insulin-stimulated glucose disposal. These observations in the muscle-specific ERα mouse are entirely consistent with those of whole body ERαΚΟ mice (Figs. 6.4 and 6.5) [146, 149]. Furthermore, additional studies by Barros et al. [141, 150] investigating alteration in GLUT4 expression in response to ovariectomy and E2 supplementation are in conflict with other published studies of similar design [132, 145, 151–153]. Given the lack of consensus ERE in the GLUT4 promoter [154] and absence of confirmatory findings in cellular reporter and chromatin immunoprecipitation assays, the regulation of GLUT4 expression by ERα requires further investigation. GLUT4 is impacted by several redundant transcriptional pathways [155, 156], and this redundancy in regulation likely underlies the maintenance of total GLUT4 transcript and protein levels in the absence of ERα as well as in humans or rodents of both genders in the context of insulin resistance, obesity, and type 2 diabetes [157–160]. Considering the concomitant increase in ERα and GLUT4 expression observed in muscle of exercise-trained humans and mice, it is conceivable that while ERα deletion fails to impact GLUT4 expression, increased ERα action may mediate in part the training-induced increase in total GLUT4 content [157, 161–166].
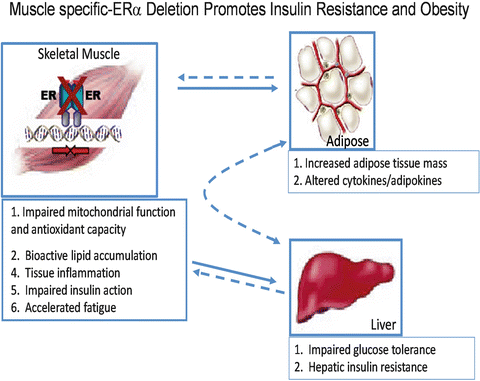
Fig. 6.5
The impact of skeletal muscle-specific ERα deletion on insulin sensitivity and adiposity in female mice (Ribas et al. [149])
Myocyte enhancer factor 2 (MEF2) expression and a functional MEF2 element in the GLUT4 promoter are critical for GLUT4 gene expression [167]. Furthermore, reciprocal regulation between ERα and MEF2 can be observed in cardiomyocytes via ERα interaction with class II HDAC [168]. Despite complex transcriptional signal integration in the regulation of GLUT4 expression [155, 156, 169–172], it is reasonable to speculate that elevated ERα action could promote increased GLUT4 transcription via heightened protein tethering with MEF2 on the GLUT4 promoter or by indirect action via AMPK [128, 173]. It is important to note that transcriptional activity of the GLUT4 promoter is quite low under basal conditions and other ovarian hormones, e.g. progesterone, are shown to play an antagonistic role in the regulation of GLUT4 expression [126]. These issues as well as the dose of E2 administration during interventional studies (presumably off-target effects of superphysiological doses of E2 are deleterious to metabolism), the age and hormone status of the human subjects and rodents used are important considerations when interpreting the literature. Given the varying roles that muscle and adipose tissue play in controlling whole body metabolic homeostasis, it is likely that the interplay of transcriptional regulators of GLUT4 vary markedly between tissues. Taken together, these data would suggest a potential role for ERα as an enhancer of GLUT4 transcription in muscle under certain conditions, but not necessarily obligatory in the direct regulation of GLUT4 expression under basal conditions.
Collectively, work by Ribas et al. suggests that the skeletal muscle insulin resistance observed in whole body ERαKO mice and animals with a muscle-specific deletion of ERα is predominantly the result of impaired insulin signal transduction [149]. A role for ERα in the regulation of proximal insulin signal transduction has been suggested previously as E2 administration to insulin resistant rodents increases insulin receptor substrate (IRS)-1 abundance and insulin-stimulated tyrosine phosphorylation and as well as the phosphotylation of Akt at Ser473 [147, 174]. Akt serves many functions in myocytes including ERα-induced regulation of myogenic differentiation [175], suppression of muscle-atrophy associated ubiquitin ligases via FOXO1 inhibition [176], and induction of genes associated with myocellular proliferation [175, 177–180]. In breast cancer cell lines, endothelial cells, and cortical neurons, ERα-specific binding and activation of PI3kinase as well as suppression of the tumor suppressor and PI3kinase inhibitory protein, PTEN, is well-established [181–185]; however, studies on these direct interactions in skeletal muscle are limited. Additionally, E2 acting via ERα is also shown to promote phosphorylation of p38 MAPK [186, 187], a signaling cascade shown to enhance GLUT4 intrinsic activity and glucose uptake [188–190]. Furthermore, it is possible that ERα activation of Akt and MAPK pathways may underlie in large part the E2-mediated protection of muscle against age-induced sarcopenia [191–197], exercise-induced muscle damage [179, 193, 198, 199], and myocyte apoptosis in the face of a variety of cellular perturbations [200–203]. Thus, ERα stimulation of muscle growth and insulin sensitivity via direct and indirect interaction with these pathways requires further exploration.
In contrast to the protective effects of ERα, it is suggested that ERβ may promote insulin resistance in skeletal muscle when the ERβ:ERα is elevated. In OVX mice, while ERα-specific activation by PPT improved muscle insulin action [129], conversely, ligand-specific activation of ERβ by DPN failed to ameliorate insulin resistance [129]. Moreover, ovariectomy of hyperestrogenic female ERα-deficient mice was shown to improve glucose tolerance and insulin sensitivity presumably though elimination of ERβ activation by endogenous Ε2 [204]. Similarly, administration of an ERβ-selective agonist to male E2-deficient ArKO mice decreased glucose uptake [150]. Finally, evidence indicates that ERβ-deficiency protects against diet-induced insulin resistance in male mice by increasing PPARγ signaling in adipocytes, which indirectly improves skeletal muscle insulin action by promoting lipid accumulation in adipose tissue and diminishing ectopic lipid deposition in muscle [205, 206]. A role for ERβ in the pathogenesis of human insulin resistance remains unknown and there is still much work to do in determining the tissue-specific interactions of these transcription factors under more physiological conditions.
ERα and Skeletal Muscle Fatty Acid Metabolism and Inflammation
Normally cycling women are protected against acute lipid-induced insulin resistance compared with estrogen-deficient women and men [120, 207]. Furthermore, muscle from premenopausal women shows enhanced insulin sensitivity despite 47 % higher triglyceride content compared with age-matched men [158]. These observations are consistent with a reduced respiratory quotient and greater reliance on the oxidation of fatty acids as a fuel source in women [208]. Interesting similarities between E2 replete women and exercise trained subjects including elevated muscle ERα expression [144, 166, 209], heightened insulin sensitivity [210], elevated muscle lipid tolerance [211], and enhanced oxidative capacity [212, 213] are consistently observed. Estrogen supplementation is shown to enhance lipid oxidation in men during acute endurance exercise [214] as well as palmitate oxidation in myotubes from male subjects ex vivo [132]. The effect of E2 to increase the expression of fatty acid transport protein FAT/CD36 and FABP as well as transcription factors and key enzymes that regulate oxidative metabolism [151, 157, 215] likely underlie these observations in male subjects. In addition, exercise and E2 are shown to rapidly stimulate AMPK phosphorylation in both muscle and myotubes [128, 216]. AMPK is considered a central regulator of many cellular processes including growth, mitochondrial biogenesis, and oxidative metabolism [217, 218]. Similar to the effects of E2, the ERα-selective agonist PPT stimulates AMPK phosphorylation in muscle of female rats [129] while OVX or whole body ERα deletion is associated with reduced skeletal muscle levels of phosphorylated AMPK [146, 219]. Muscle PPARα, PPARδ, and UCP2 expression are also reduced in whole body ERαKO mice suggesting that ERα is essential in the regulation of a coordinated program regulating oxidative metabolism. Importantly, although the impairment in muscle fatty oxidation was recapitulated in the muscle-specific ERαKO mice (MERKO), no alteration in basal p-AMPK, PPARα, PPARδ, or UCP2 was observed [149], thus suggesting that these specific alterations in muscle gene expression in ERαKO mice are secondary to the loss of ERα in other metabolic tissues, likely the CNS, adipose, or liver. Despite these model differences, the skeletal muscle insulin resistance and bioactive lipid accumulation (triacylglycerol, diacylglycerol, and ceramides) was surprisingly similar between ERαKO and MERKO. Consistent with these observations, oxygen consumption rates in C2C12 myotubes with ERα knockdown were reduced significantly. In addition, mitochondria from muscle cells depleted of ERα produced high levels of reactive oxygen species thus precipitating increased cellular oxidative stress. Collectively these data support the notion that ERα is critical for maintaining fatty acid oxidation in skeletal muscle by mechanisms including the regulation of: (1) fatty acid transport into the cell, (2) activation of intermediary signaling critical for shifting substrate metabolism, (3) transcriptional regulators of fatty acid metabolism, and mitochondrial function. Thus, ERα expression in skeletal muscle may be a central regulator of adiposity by indirect action as MERKO mice reproduced the obesity phenotype observed in the whole body ERαKO (Fig. 6.5).
Moreover, E2 treatment reduces HFD-induced insulin resistance in skeletal muscle by 50 % (assessed by hyperinsulinemic-euglycemic clamp) in an ERα-dependent manner [147]. The mechanistic link between the accumulation of lipid intermediates, activation of inflammatory signaling cascades, and impaired insulin action is shown in myocytes and rodent muscle, and indeed these factors are observed concurrently in obese, type 2 diabetic subjects [220–223], as well as muscle from whole body and muscle-specific ERαKO mice [146]. Bioactive lipid intermediates including diacylglycerol and ceramides are believed to activate stress kinases including IKKβ, c-Jun-N-terminal kinase (JNK), and certain nPKCs [221, 224–226]. Indeed, muscle from normal chow-fed whole body ERαKO mice showed heightened inflammatory signaling as reflected by markedly increased JNK phosphorylation and TNFα transcript [146]. In addition to the increase in bioactive lipid intermediates found in ERαKO muscle, the production of reactive oxygen species as well as the possible ERα de-repression of selective inflammatory targets within the nucleus are likely mediators of heightened muscle inflammation.
Markers of inflammation and oxidative stress are elevated in rodent models of obesity and in patients with type 2 diabetes [227, 228]. Myotubes and skeletal muscle with ERα deletion showed a marked reduction in Gpx3 expression, an antioxidant enzyme reported to scavenge hydrogen peroxide and diminish oxidative stress [145, 146]. E2 replacement in OVX animals also increased Gpx3 expression in skeletal muscle [145]. Given that Gpx3 expression levels in skeletal muscle are elevated in females compared to male [229], reduced in T2DM patients [230], are associated with insulin resistance and metabolic dysfunction [230], and the gene is now identified as a causal candidate for obesity [231], additional work studying the direct role of estrogen action in the regulation of anti-oxidant enzymes appears warranted.
Although reductions in mitochondrial number and function have been implicated in the pathobiology of insulin resistance [232–235], and indeed gender dimorphisms in mitochondrial biology have been described [236], whether E2/ERα preserves insulin action by maintenance of mitochondrial integrity remains unknown. Emerging unpublished findings from the Hevener laboratory indicate that skeletal muscle ERα is critical for the maintenance of mitochondrial function and the turnover of damaged organelles. However, the mechanisms underlying these observations remain incompletely understood at the present time.
ERs and Hepatic Insulin Sensitivity
Hepatic insulin resistance contributes to impaired glucose tolerance and fasting hyperglycemia of type 2 diabetes by unrestrained hepatic glucose production. Although a direct role of the liver in the insulin resistance phenotype induced by E2 deficiency or tissue-selective ablation of ERα remains unclear. Bryzgalova et al. showed that the global insulin resistance of female mice with a homozygous ERα null mutation was due almost exclusively to impaired suppression of hepatic glucose production (HGP) during euglycemic-hyperinsulinemic clamp studies in anesthetized mice [148]. In contrast, Ribas et al., reported in the same ERα-deficient female mice only modest reduction in liver insulin sensitivity during euglycemic-hyperinsulinemic clamp studies in conscious animals [31]. Thus, the possibility exists that anesthesia artificially contributed to the severe hepatic insulin resistance phenotype observed by Bryzgalova et al. in ERα-deficient mice [148]. Studies in liver-specific ERα-deficient mice should provide the definitive findings required to determine the role of liver ERα in controlling hepatic insulin action and glucose tolerance. Still, E2 and PPT treatments ameliorate insulin resistance in genetically obese leptin resistant and high fat diet-fed mice [147, 237]. Numerous studies are in agreement that E2 suppresses lipogenic gene expression, triglyceride accumulation, and steatosis in liver of HFD-fed [237] and leptin-resistant female mice (Fig. 6.6) [238]. Interestingly, this effect is not always reproduced by the ERα, selective agonists PPT [239]. A significant limitation of studies inducing chronic estradiol elevation is the inability to ascribe specific actions of estradiol or ERα agonism to a given tissue as circulating estradiol impacts all metabolic tissues producing numerous secondary phenotypes due to extensive tissue crosstalk. Moreover, E2, and possibly ERα, cyclicity appears critical in the regulation of gene expression and cellular function, and this cyclicity is eliminated during chronic E2 or ERα agonist administration. Together, these data suggest that ERα activation protects from hepatic insulin resistance by preventing ectopic lipid accumulation in liver (lipotoxicity), but the direct involvement of ERα and the molecular mechanisms of action in hepatocytes require further clarification.
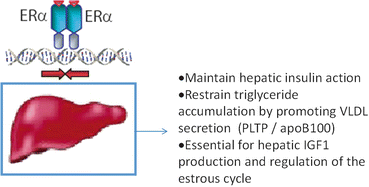
Fig. 6.6
Proposed actions of estrogen and ERα in the protection against hepatic triglyceride accumulation and insulin resistance
Estrogen Action, Immunity, and the Metabolic Syndrome
Estrogens affect many immune and inflammatory conditions including auto-immune diseases [240–245] as well as immuno-modulatory responses to parasitic and bacterial infection [246–251]. Following OVX, immune cell infiltration and increased tissue inflammation (TNFa, iNOS, and CD11c) coincided with a ~4 fold increase in perigonadal and inguinal fat. The T-cell marker CD3 and the Th1 cytokine interferon γ were also elevated in perigonadal fat from ovariectomized female mice [32] suggesting that the absence of E2 promotes immune cell inflammation. Indeed, similar to findings in rodents, circulating levels of pro-inflammatory cytokines are elevated in women following natural or surgical menopause [124]. In line with these studies, work by the laboratory of Pierre Gourdy showed that E2 heightens the inflammatory response to intraperitoneal injection of thioglycollate or lipopolysaccharide, and that ERα is critical in mediating these actions as well as reducing bacterial burden through phagocytosis [248]. Taken together, these data suggest that ERα expression in immune cells is critical for mediating a variety of cellular responses necessary for normal innate and adaptive immunity.
ERα is expressed in macrophages and other immune cells known to exert dramatic effects on glucose homeostasis. Macrophages are central effector cells of innate and adaptive immunity, and over the past decade their role in modulating whole body metabolism and insulin sensitivity has taken on increasing interest [252, 253]. Recently Ribas et al. investigated the impact of ERα expression on macrophage function to determine whether hematopoietic or myeloid-specific ERα deletion manifests an obesity-induced insulin resistance phenotype in mice [254]. This group sought to determine how much of the whole body ERαKO phenotype could be recapitulated by deletion of ERα specifically from myeloid cells. Indeed, altered plasma adipokine and cytokine levels, glucose intolerance, insulin resistance, and increased adipose tissue mass were observed in animals harboring a hematopoietic or myeloid-specific deletion of ERα (Fig. 6.7) [254]. A similar obese phenotype with increased atherosclerotic lesions was produced in LDLR-KO mice transplanted with ERαKO bone marrow. In isolated macrophages, Ribas et al. found that ERα is necessary for repression of inflammation, maintenance of oxidative metabolism, IL4-mediated induction of alternative activation, full phagocytic capacity in response to LPS, and oxidized LDL-induced expression of ApoE and Abca1 (Fig. 6.7) (301). Moreover, bone marrow-derived macrophages lacking ERα secrete factors that induce skeletal muscle and adipocyte insulin resistance in culture [254]. A major limitation in the field is the failed identification of these pro-inflammatory insulin-resistance producing substance secreted from immune cells. Thus, metabolomic and proteomic analyses will be necessary to move the field forward in this regard. It is likely that these macrophage-secreted factors include a combination of cytokines, fatty acids, and reactive oxygen/nitrogen species that act to alter metabolic function of adjacent cells in contact or in close proximity with tissue resident macrophages.
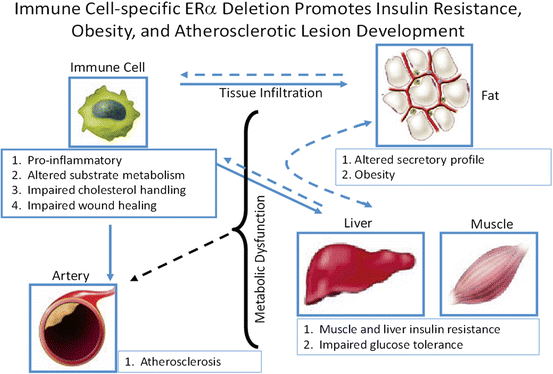
Fig. 6.7
 The Role of Estrogens in the Regulation of Peripheral Glucose Dynamics
The Role of Estrogens in the Regulation of Peripheral Glucose Dynamics
 Influence of Ovarian Hormones on Skeletal Muscle Contractility
Influence of Ovarian Hormones on Skeletal Muscle Contractility
 Transitions Across a Lifetime: Unique Cardiovascular Physiology of Women and Relationship to Cardiovascular Disease Risk
Transitions Across a Lifetime: Unique Cardiovascular Physiology of Women and Relationship to Cardiovascular Disease Risk
 Metabolic Health in the Aging Female: Human Perspective
Metabolic Health in the Aging Female: Human Perspective
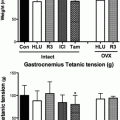 Estrogen Effects on Skeletal Muscle
Estrogen Effects on Skeletal Muscle
 The Contribution of Ovarian Hormones to the Cellular Regulation of Lipid Metabolism
The Contribution of Ovarian Hormones to the Cellular Regulation of Lipid Metabolism




Myeloid-specific ERα deletion promotes obesity, insulin resistance, and atherosclerosis susceptibility in female mice
Related posts:
The Role of Estrogens in the Regulation of Peripheral Glucose Dynamics
Influence of Ovarian Hormones on Skeletal Muscle Contractility
Transitions Across a Lifetime: Unique Cardiovascular Physiology of Women and Relationship to Cardiovascular Disease Risk
Metabolic Health in the Aging Female: Human Perspective
Estrogen Effects on Skeletal Muscle
The Contribution of Ovarian Hormones to the Cellular Regulation of Lipid Metabolism
Stay updated, free articles. Join our Telegram channel
Full access? Get Clinical Tree
