Alzheimer’s disease (AD)
An estimated 5.2 million Americans have AD—including 5 million aged 65 and older or one in nine people aged 65 and older.
Of those aged 75 or older, 44 % have it. One third (32 %) of those aged 85 and older have AD. Approximately 200,000 under age 65 have early onset AD
Vascular dementia (VD)
VD is the second most common cause of dementia after Alzheimer’s disease, and is caused by “mini strokes” or occlusions of blood vessels in the brain.
VD “paves the way” for Alzheimer’s disease to develop, and VD/ALZ is a frequent diagnosis
Dementia with Lewy bodies (DLB)
People with Lewy body dementia often have memory loss and thinking problems similar to that seen in AD. However, the initial presentation of DLB includes sleep disturbances, hallucinations, and muscle rigidity or other movements that mimic Parkinson’s disease.
The brain changes of DLB alone can cause dementia, or these brain changes can coexist with the brain changes of Alzheimer’s disease and/or vascular dementia, with each abnormality contributing to the development of dementia. When this happens, the individual is said to have “mixed dementia”
Parkinson’s disease (PD)
As PD progresses, it often results in a progressive dementia similar to DLB or AD. Symptoms include problems with movement early on in the disease. If dementia develops, symptoms are often similar to DLB.
The brain changes involve alpha-synuclein clumps that develop deep in the brain in an area called the substantia nigra. These clumps are thought to cause degeneration of the nerve cells that produce dopamine
Frontotemporal dementia (FTD)
Typical symptoms of FTD include changes in personality and behavior and difficulty with language. Nerve cells in the front and side regions of the brain are especially affected.
Some people with FTD lose empathy for others as well as a sense of “what is appropriate” to say and do in public settings. Others lose language skills.
There are no distinguishing microscopic abnormalities linked to all cases. People with FTD generally develop symptoms at a younger age (at about age 60) and survive for fewer years than those with AD
Creutzfeldt–Jakob disease (CJD)
Prion diseases such as CJD occur when prion protein, which is found throughout the body but whose normal function is not yet known, begins folding into an abnormal three-dimensional shape. CJD develops when prion protein in the brain also begins to fold into the same abnormal shape
Through a process scientists do not yet understand, misfolded prion protein destroys brain cells. Resulting damage leads to rapid decline in thinking and reasoning as well as involuntary muscle movements, confusion, difficulty walking, and mood changes. Sign up for our enews to receive updates about Alzheimer’s and dementia care and research
CJD is rare, occurring in about one in 1 million people annually worldwide
Experts generally recognize the following main types of CJD:
Sporadic CJD develops spontaneously for no known reason. It accounts for 85 % of cases. On average, sporadic CJD first appears between ages 60 and 65
Familial CJD is a heredity form caused by certain changes in the prion protein gene. These genetic changes are “dominant,” meaning that anyone who inherits a CJD gene from an affected parent will also develop the disorder. Familial CJD accounts for about 10–15 % of cases
Infectious CJD is an especially rare form of CJD and results from exposure to an external source of abnormal prion protein.
These sources are estimated to account for about 1 % of CJD cases. The two most common outside sources are:
Medical procedures involving instruments used in neurosurgery, growth hormone from human sources or certain transplanted human tissues.
The risk of CJD from medical procedures has been greatly reduced by improved sterilization techniques, new single-use instruments and synthetic sources of growth hormone
The brain’s patterns of electrical activity is similar to the way an electrocardiogram (ECG) measures the heart’s electrical activity
Brain magnetic resonance imaging (MRI) can detect certain brain changes consistent with CJD
Lumbar puncture (spinal tap) tests spinal fluid for the presence of certain proteins
Causes and risks
Sporadic Creutzfeldt–Jakob disease has no known cause. Most scientists believe the disease begins when prion protein somewhere in the brain spontaneously misfolds, triggering a “domino effect” that misfolds prion protein throughout the brain. Genetic variation in the prion protein gene may affect risk of this spontaneous misfolding
Mutations in the prion protein gene also may play a yet-to-be-determined role in making people susceptible to infectious CJD from external sources. Scientists do not yet know why infectious CJD seems to be transmitted through such a limited number of external sources. Researchers have found no evidence that the abnormal protein is commonly transmitted through sexual activity or blood transfusions
Normal pressure hydrocephalus (NPH)
Symptoms of NPH include difficulty walking, memory loss, and inability to control urination, which are caused by the buildup of fluid in the brain. NPH can sometimes be corrected with surgery
Huntington’s disease dementia (HDD)
HDD is a progressive brain disorder caused by a single defective gene on chromosome 4. Symptoms include abnormal involuntary movements, a severe decline in thinking and reasoning skills, and mood changes including irritability, depression, and others. The gene defect causes abnormalities in a brain protein that, over time, lead to worsening symptoms
Challenging Behaviors: Blame the Brain!
Another area that calls for honest discussion is the connection between challenging behaviors and specific brain damage . Why? Because without this knowledge, caregivers often arrive at the conclusion that their loved one with Alzheimer’s is just being “difficult” and deliberately so! It is much more challenging to be patient with someone who seems to have some control over his/her behavior and is choosing to act in an unkind and difficult manner than it is to be patient with someone whose behaviors are directly linked to brain damage. Knowing that the behaviors result from brain damage removes personal responsibility from the patient—the behaviors naturally flow from what is happening in the brain and the behaviors are not deliberate attempts to frustrate or “get even with” the caregiver (Swaab 2014, pp. 348–351). Armed with knowledge connecting specific brain damage to specific behaviors allows caregivers to learn strategies to respond to these behaviors. It is not necessary for caregivers to have in-depth knowledge about the brain. A superficial, yet specific knowledge of how damage is linked to challenging behaviors is enough. Let us take a look at what level of knowledge is useful to caregivers. (Carson and Smarr 2007)
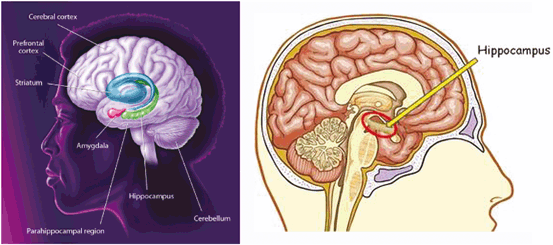
An area of the brain that is damaged very early on in Alzheimer’s is the hippocampus shown in the picture on the left. This structure processes every experience that we have and stores long-term memories—sending the short-term memories to another part of the brain. In Alzheimer’s, the brain becomes less and less able to process short-term memories, and since the “old” memories still remain, the person begins to “live” increasingly “in the past.”
By stage 6 on the functional assessment scale (FAST) scale, the person has no more than 5 min of short-term memory and can only participate in one activity at a time. The lack of short-term memory means that questions are repeatedly asked and stories are retold. This is because the person asking the questions and retelling the stories has no memory of having repeatedly asked the questions or told the same story. How can a caregiver calmly respond to this repetition? There are many ways that caregivers can make use of the person’s deficits. If the activity is repetitive, mindless, and yet productive, the caregiver might redirect the person to participate in repetitive activities that he or she enjoys.
Related posts:


Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


