
Arylsulfatase A (ARSA) mutations that result in low activity (between 5 and 20% of normal) are common in European, American and other populations but are believed not to cause “classical” clinical presentation of metachromatic leukodystrophy. They have been referred to as pseudo-deficiencies (see Chapter 9). However, genotype–phenotype analysis of these individuals suggests that ARSA pseudodeficiencies may contribute to the clinical severity of metachromatic leukodystrophy [19], multiple sclerosis [3] and possible onset of neuropsychological and non-verbal learning disabilities [20, 21].
Although mucolipidosis IV is a pan-ethnic neurodegenerative disorder, with most patients of Ashkenazi Jewish origin (see Chapter 17), its frequency may be much greater than currently appreciated because its common presentation as a cerebral palsy-like encephalopathy may lead to misdiagnosis, particularly with “‘milder” patients. The problem of accurately defining some LSD phenotypes is further illustrated by mutations (and so-called polymorphisms) in the ß-glucocerebrosidase gene, which result in Gaucher disease but are also associated with Parkinson disease. Over the coming years it is likely that many other genes/proteins will be identified as causing dysfunction of the endosome-lysosome-network with yet to be defined clinical outcomes.
Incidence and prevalence
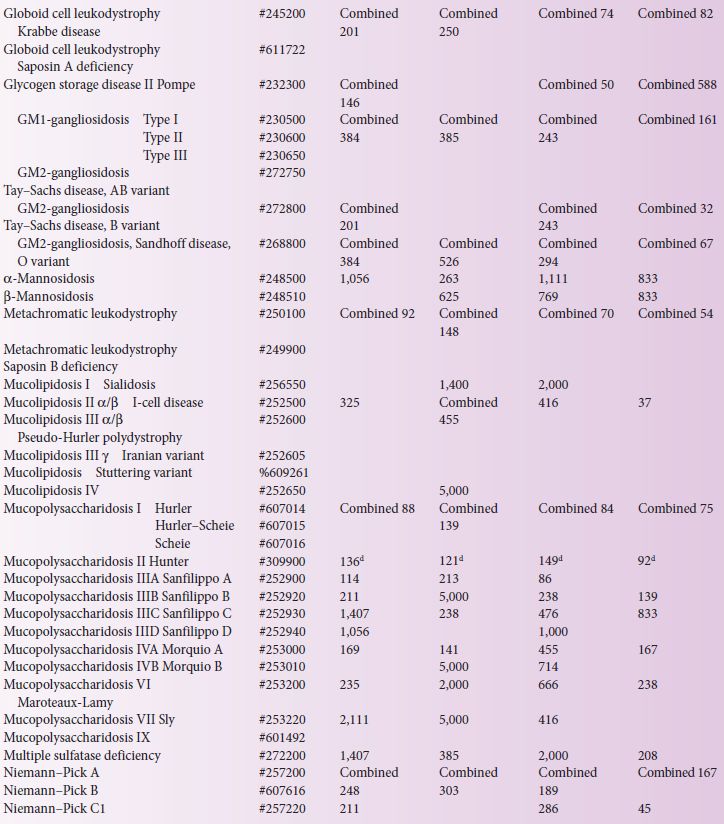
LSD incidence, defined as the total patient number divided by total births in the same period of time, has been reported in a number of countries (Table 4.1). Major difficulties associated with obtaining accurate epidemiological data involve the individual rarity of these disorders and the numerous diagnostic centres that often use different diagnostic methods. Most LSD patients are diagnosed after clinical presentation, and this is often the basis upon which to evaluate the likely genetic and biochemical tests required to fully define the LSD and its likely prognosis.
Importantly, if the studies have missed a diagnosis considerable differences may result to a calculated prevalence for an extremely rare disease; care should therefore be exercised to estimate inheritance risk from this data. Further, when comparing in Table 4.1 data that have been collected from different studies, the nature of the methods used to calculate birth prevalence and incidence values must be considered. For instance, the Australian study [8], based on clinical diagnoses between 1980 and 1996, assumed that late-onset patients who were diagnosed during this period were representative of late-onset patients born within the study dates but yet to present clinically; whereas in The Netherlands [13] the period used to calculate birth prevalence was based on the patient’s age, with the assumption that patients born prior to the last patient in the subgroup were identified. Both methods have limitations and it is likely that the identification of late-onset patients has been underestimated. In Australia, Pompe disease has a clinical incidence of 1 in 146,000 births, whereas, based on carrier detection studies in the USA and The Netherlands, an incidence as high as 1 in 40,000 is likely [2, 7], suggesting that adult Pompe patients may be under-detected in the Australian population.
In recent times the observed discrepancy between the measured LSD incidence and the predicted carrier rates suggests that LSD cases are substantially under-diagnosed in the general population. For instance, an under-diagnosis rate may be particularly high among individuals with LSD gene mutations that lead to disease characterized by psychotic symptoms or stroke and cardiac problems. Newborn screening for LSD has identified much higher incidences of some LSDs – for example, Fabry disease – than previously estimated based on classical Fabry syndrome presentation alone (see “Populations at a high-risk” and “Population screening and diagnostic methods” below and Chapter 23). Thus, the LSD overall (and specific disease) incidence may be much higher than currently appreciated.
Retrospective data collected in Australia (from 1980 to 1996) reported 470 diagnoses representing 27 different LSDs (Table 4.1). The incidence ranged from 1 in 57,000 for Gaucher to 1 in 4,222,000 for Mucolipidosis type I. The combined incidence of all LSDs, with prenatal diagnoses included, was 1 in 7,700 births [8].
In The Netherlands, 963 LSD patients were diagnosed between 1970 and 1996, giving an incidence of 1 in 7,100 births, with glycogen storage disease II the most prevalent at 1 in 50,000 [13]. Between 1975 and 2008, 478 LSD patients were reported in the Czech Republic with 34 different LSDs and an overall LSD prevalence of 1 in 8,000 births, with Gaucher being most prevalent at 1 in 89,000 [14]. In Northern Portugal, 222 LSD patients were identified over approximately 20 years, giving an incidence of 1 in 2,500; 29 different LSD patients were reported with GM2-gangliosidosis, Niemann–Pick C and Gaucher being the most prevalent at 1 in 31,950, 45,450 and 74,070, respectively [12].
Populations at a high-risk
Related posts:





Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


