Prolymphocytic Leukemia
The Royal Marsden NHS Foundation Trust, Sutton and Institute of Cancer Research, London, UK
1. What are the characteristic clinical features of PLL?
PLL usually presents in the sixth decade or older and occurs more frequently in males. Characteristically, patients have rapidly increasing lymphocyte counts and splenomegaly. Low-volume lymphadenopathy, skin rashes, peripheral edema, and pleuro-peritoneal effusions are seen relatively frequently in T-cell PLL (T-PLL) but not in B-cell PLL (B-PLL). Central nervous system (CNS) involvement has been described in both, but is rare. A minority of patients may be asymptomatic at diagnosis, and this “‘indolent” phase can persist for a variable length of time. However, progression can be very rapid when it occurs, and patients should therefore be monitored closely (every 1–3 months depending on the rate of change). Clinical characteristics of PLL are summarized in Table 33.1.
Table 33.1 Clinical and laboratory characteristics of B- and T-cell prolymphocytic leukemias.
| Characteristic findings | B-PLL | T-PLL |
|---|---|---|
| Clinical features | Median age: 69 M:F ratio: 1.6:1 B-symptoms Splenomegaly Minimal lymphadenopathy High WBC | Median age: 61 M:F ratio: 2:1 Splenomegaly, lymphadenopathy, skin rash, edema, and pleuro-peritoneal effusions Very high WBC |
| Morphology | >55% prolymphocytes (usually >90%) Prolymphocyte is 2× size of CLL lymphocyte | Basophilic prolymphocytes with cytoplasmic blebs Small-cell (20%) and SS (5%) variants |
| Immunophenotype | SmIG strong, CD19+, CD20+, CD22+, CD79a+, CD23−, CD5−/+ FMC7+ (CLL score 0–1) | CD2+, CD3+, CD5+, CD7++ CD4/8 variable CD1a−, TdT−, CD25−/+ |
| Cytogenetics | 13q del, 11q del, 17p del, 6qdel No t(11;14) | t(14;14); inversion 14; t(X;14); iso8q; complex |
| Oncogenes | TP53, C-MYC | TCL1, MTCP1, ATM |
| Differential diagnosis | T-PLL, CLL/PL, MCL (leukemic phase), SMZL, HCL-v | B-PLL, T-LGL leukemia, A-TLL, SS |
| Prognosis | Median survival: 3 years | Median survival: 7 months with conventional therapy; 20 months with alemtuzumab; 37 months with alemtuzumab + HSCT |
A-TLL, Adult T-cell leukemia lymphoma; B-PLL, B-cell prolymphocytic leukemia; CLL/PL, chronic lymphocytic leukemia with increased prolymphocytes (<55%); HCL-v, hairy cell leukemia variant; HSCT, hematopoietic stem cell transplant; MCL, mantle cell lymphoma; SMZL, splenic marginal zone lymphoma; SS, Sézary syndrome; T-LGL, T large granular lymphocytic leukemia; T-PLL, T-cell prolymphocytic leukemia; WBC, white blood cell count.
2. Are there any predisposing conditions?
Rarely, the diagnosis of T-PLL is made in a patient with a preceding history of an inherited genetic disorder such as ataxia telangiectasia (AT) or Nijmegen breakage syndrome. There are no other familial or geographical predisposing features.
3. What does the peripheral blood look like?
Prolymphocytes are medium-sized cells (twice the size of a chronic lymphocytic leukemia (CLL) lymphocyte) with a high nuclear-to-cytoplasmic ratio, a single prominent nucleolus, and basophilic agranular cytoplasm. In B-PLL, prolymphocytes should account for more than 55% of peripheral blood lymphoid cells, and the proportion usually exceeds 90% (Figure 33.1A). No cytoplasmic hairy projections or “villi” are seen in B-PLL in contrast to hairy cell leukemia variant (HCL-v) and splenic marginal zone lymphoma (SMZL). In B-PLL and in 50% of T-PLL cases, the cells have a round to oval nucleus. In the remainder of T-PLL cases, the nuclei are irregular, often with convolutions, although they are less pronounced than those seen in Sézary or adult T-cell leukemia/lymphoma (ATLL) cells. In typical T-prolymphocytes, the cytoplasm is more intensely basophilic and has an irregular outline with “blebs” (Figure 33.1B). Two variants, small cell (previously known as T-CLL, a term no longer used) and cerebriform, are seen in approximately 20% of cases. Both these variants have immunophenotypic, cytogenetic, and clinical features that are similar to those of typical T-PLL.
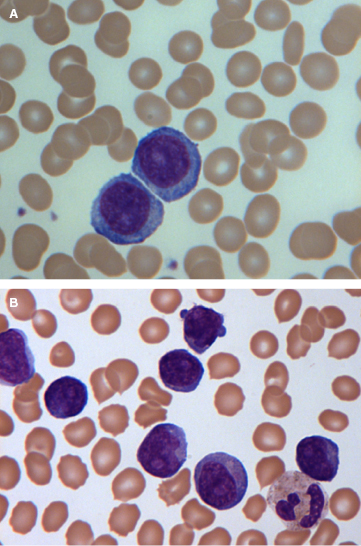
4. Is examination of the BM or other histology helpful?
In B-PLL, BM, lymph node, and spleen histology may all be important in confirming the diagnosis and distinguishing this from other B-cell disorders, whilst in T-PLL these are rarely needed.
5. How can B- and T-cell PLL be distinguished?
Despite the clinical and morphological similarities, the B- and T-cell subtypes of PLL can be distinguished readily by immunological markers. The laboratory features of PLL are summarized in Table 33.1. In B-PLL, the monoclonal B-cell proliferation is confirmed by establishing light-chain restriction, and the B-cells are further characterized by use of a panel of immunophenotypic reagents. Importantly, this will rule out the presence of typical CLL (CD5+, CD23+, weak surface immunoglobulin, and CD79b) or CLL with an increase in prolymphocytes (CLL/PL), which has the same phenotype. In contrast, in B-PLL there is strong Ig and CD79b expression, and most cases are CD23− and CD5− negative. T-prolymphocytes have a postthymic (TdT− and CD1a−) T-cell phenotype (CD5+, CD2+, and CD7+) (Figure 33.2), with variable expression of CD4 and CD8. Not all cases will express membrane CD3, although this is invariably present in the cytoplasm, and CD7 expression is strong in contrast to other mature T-cell leukemias, where this marker is often weakly positive or negative. CD25, CD38, and class II HLA-DR may be variably expressed, but markers identifying cytotoxic T-cells such as TIA-1 are negative, even in cases with a CD8+ phenotype.
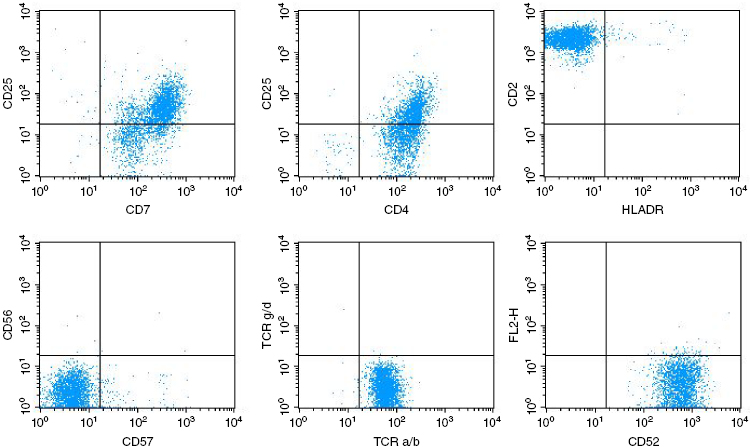
6. What other specialist tests are useful?
Cytogenetics, and in some cases molecular tests, can be helpful in confirming the diagnosis. The commonest cytogenetic abnormality seen in B-PLL is del 17p involving loss of TP53. Importantly, t(11,14), which is the hallmark translocation for mantle cell lymphoma (MCL), is not seen. Rarely there are translocations involving C-MYC. The majority of T-PLL cases will have complex karyotypes, typically with abnormalities involving chromosome 14 (Figure 33.3), and frequently also chromosomes 8 and 11. These changes result in the activation of oncogenes (TCL-1, MTCP-1, and ATM) that are involved in the pathogenesis of this disorder.
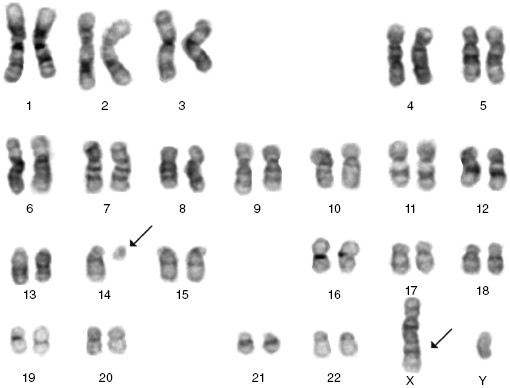
7. What is the prognosis for patients with PLL?
There is little information regarding prognostic factors in PLL. Patients will often be aware from their reading of the literature that this is an aggressive leukemia with poor overall survival. However, this information is largely based on retrospective data, and the outlook has improved with the introduction of newer treatment approaches.
In B-PLL, retrospective data suggest a median survival of only 3 years. The presence of TP53 deletions or mutations predicts for poor response to conventional treatment and shorter survival. Very little information is available from prospective series in the era of antibody therapy that, as in CLL, may have resulted in significant improvement in survival.
In our data set for T-PLL, biological parameters such as immunophenotype, cytogenetics, and molecular genetics do not influence survival or response to therapy. The most important predictor of outcome is response to alemtuzumab therapy. In this regard, patients with extramedullary disease (e.g., liver, CNS, and pleuro-peritoneal effusions) have lower response rates (RR) to alemtuzumab. In our series of 86 patients treated with alemtuzumab, nonresponders had a median survival of only 4 months. However, even in some older patients (over 80 years), survival has exceeded 5 years following single-agent alemtuzumab, and in some younger patients who have undergone hematopoietic stem cell transplantation (HSCT), remissions have exceeded 10 years. In the only other large series reported from the MD Anderson Cancer Center (MDACC), 5-year survival from diagnosis was 21% and poorer outcome was associated with high WBC, short lymphocyte doubling time, older age, and high expression of TCL-1 protein measured by flow cytometry and immunohistochemistry.
In the future, there is some hope that survival will improve further in both B- and T-cell PLL with the availability of additional effective therapies.
8. When should treatment for PLL be started?
The majority of patients with PLL will require treatment immediately for symptomatic disease. In those patients presenting with an asymptomatic lymphocytosis that is relatively stable or only slowly progressive, it is possible to monitor until evidence of disease progression. However, the progression may occur rapidly, and therefore closer monitoring is required than would be the case for patients with early-stage CLL. This will often allow time to identify a suitable donor if allogeneic transplant is planned, so that there will be no delays once treatment is initiated. The duration of an indolent phase is variable, but it is unusual for it to persist for more than 1–2 years.
9. What is the best first-line therapy for PLL?
Related posts:
 17: Management of Therapy-Related Myeloid Neoplasms
32: Hematopoietic Cell Transplantation in Chronic Lymphocytic Leukemia
17: Management of Therapy-Related Myeloid Neoplasms
32: Hematopoietic Cell Transplantation in Chronic Lymphocytic Leukemia
 36: Nodular Lymphocyte-Predominant Hodgkin Lymphoma
82: Preoperative Systemic Therapy for Breast Cancer
109: Surgical Aspects of Upper Gastrointestinal Cancers
36: Nodular Lymphocyte-Predominant Hodgkin Lymphoma
82: Preoperative Systemic Therapy for Breast Cancer
109: Surgical Aspects of Upper Gastrointestinal Cancers
 34: Hairy Cell Leukemia
34: Hairy Cell Leukemia



