Laboratory diagnosis
Measurement of cystine levels in polymorphonuclear leukocytes and cultured fibroblasts is typically used for diagnosis. The prenatal diagnosis of cystinosis is based genetic analyses or on the determination of increased amount of free-cystine present in amniotic fluid cells.
Sialic acid storage disease
Genetic basis
Mutations in the SLC17A5 (solute carrier family 17 (anion/sugar transporter), member 5) gene, which is localized on chromosome 6q14–15, cause the different forms of sialic acid storage disease (SASD, Online Mendelian Inheritance in Man #604369, #269920). The gene product, sialin, transports free sialic acid out of the lysosomes. Sialin can also transport glucuronic and iduronic acids across the lysosomal membrane. Approximately 20 mutations have been identified in the SLC17A5 gene. The majority of patients are from Finland, Sweden or the Netherlands. These mutations result in loss of synthesis of the protein or defective transport of the protein to the lysosomal membrane.
Pathophysiology.
Mutations in the SLC17A5 gene that reduce or eliminate sialin transport activity result in an accumulation of the acidic monosaccharide, sialic acid, in lysosomes, which increase in number and become swollen. It is still unexplained how this defect in the transport of sialic acid leads to a brain developmental defect and to neurodegeneration.
Clinical presentation.
Sialic acid storage disease [6] primarily affects the nervous system. Patients with sialic acid storage disease have signs and symptoms that may vary widely in severity. Infantile free sialic acid storage disease (ISSD) is the most severe form of this disorder. Severe developmental delay, coarse face, hypotonia, failure to thrive, the development of ascites, nephrosis, seizures, multiple dysostosis, hepatosplenomegaly, and cardiomegaly are hallmarks of ISSD. Affected infants may have foetal hydrops and hydrocephalus. Children with this severe form of the condition usually live only into early childhood. Salla disease is a less severe form of sialic acid storage disease, which leads to hypotonia during the first year of life and progressive neurological symptoms, intellectual disability, developmental delay, seizures, ataxia, spasticity and athetosis. Individuals with Salla disease usually survive into adulthood. People with a phenotype intermediate between that of classic Salla disease and ISSD have been described.
Laboratory diagnosis
The increased excretion of free sialic acid in the urine forms the basis of a screening test for SASD. Diagnosis is confirmed by quantitative measurement of of free sialic acid in the patient´s urine or fibroblasts and by mutation analysis of the sialin gene. Prenatal diagnosis is available for pregnancies known to be at risk by measurement of free sialic acid in amniotic fluid supernatant (AFS) or mutation analysis. The measurement of free sialic acid in AFS is also used to screen for SASD in cases of foetal hydrops. Prenatal diagnosis of a new case of Salla disease can be made by measuring the amount of sialic acid in fetal cells followed preferably by genetic testing.
Danon disease
Genetic basis
LAMP2, which is located at Xq24, encodes for one of the most abundant lysosomal membrane proteins LAMP-2, which can be expressed in three isoforms (LAMP-2A, LAMP-2B and LAMP2-C). Danon Disease (Online Mendelian Inheritance in Man #300257) is caused by premature truncation of the LAMP-2 protein [7]. About 35 different mutations (frameshifts, stopcodons, and splicing site mutations) over the whole gene have been described to date. Most mutations are private, being reported in just a single family with the notable exceptions of recurrent mutations in exons 3 and 7.
Pathophysiology
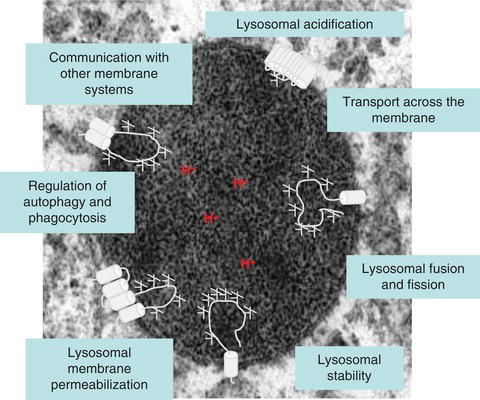
The disease manifests as a serious skeletal and cardiac myopathy associated with morbidity and early mortality. A hallmark is the accumulation of autophagic vacuoles with sarcolemnal features in myofibres. LAMP-2 has been postulated to control the autophagic flux, lysosomal motility and lysosomal degradation of certain cytoplasmic proteins. All three known LAMP-2 isoforms (LAMP-2-A, B and C) have the same large luminal domain whereas the transmembrane and short cytosolic domains differ. Whereas the LAMP-2A isoform has been implicated in the direct transport of cytosolic proteins into the lysosome (chaperone-mediated autophagy) the LAMP-2B isoform seems to be more important for regulating macroautophagy.
Clinical presentation
Related posts:





Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


