Management of Myelodysplastic Syndromes
1University of Maryland Greenebaum Cancer Center, Baltimore, MD, USA
2H. Lee Moffitt Cancer Center, Tampa, FL, USA
1. What is the general approach to the treatment of newly diagnosed myelodysplastic syndrome (MDS)?
Therapy should be tailored to each patient according to the specific risk profile, and whenever possible, patients should be treated on a clinical trial. Chapter 15 has defined the prognostic stratification in MDS. A general overview of treatment is summarized in Figure 16.1.
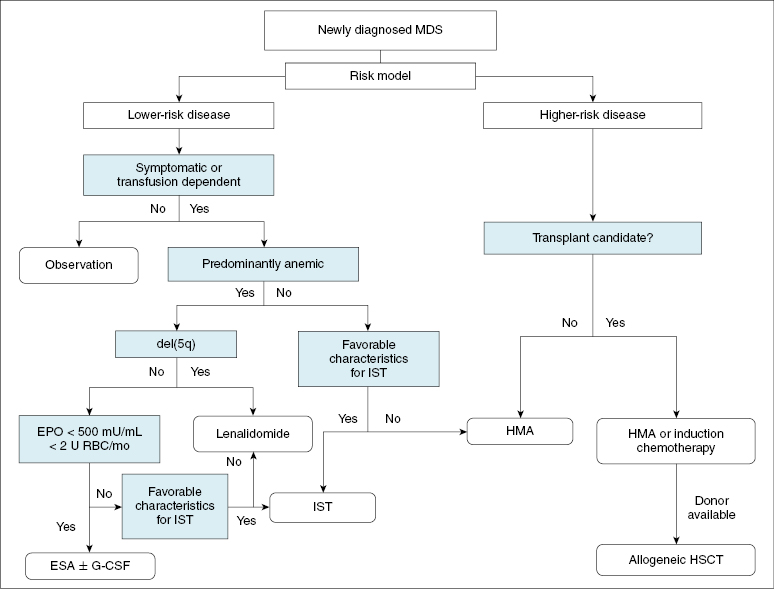
2. Which patients are good candidates for treatment with erythropoiesis-stimulating agents (ESAs) such as epoetin alpha or darbepoietin?
ESAs are an appropriate initial treatment for anemia in lower-risk MDS patients. A simple, validated decision model was developed by the Nordic MDS Group for the use of epoetin alpha and granulocyte colony-stimulating factor (G-CSF) in patients with lower-risk MDS based on their pretreatment serum erythropoietin (EPO) level and the number of red blood cell (RBC) transfusions administered each month. Patients are assigned +2, +1, and −3 points for EPO levels of <100, 100–500, or >500 U/L, respectively. Similarly, patients who require fewer than two units of packed RBCs (pRBCs) each month are assigned +2 points, and those who require at least two units each month are assigned −2 points. Patients with a combined score that was greater than +1 had a 74% probability of response, compared to 23% for those with a score between −1 and +1 and 7% for those with scores less than −1. Darbepoetin appears to be equivalent to epoietin alpha, and the use of either agent is reasonable. We typically recommend a 6–8-week trial of ESAs, followed by continuation of treatment in responders (the median duration of response is 12–18 months). We reserve the addition of G-CSF for patients who do not respond after a 6–8-week trial of an ESA, a method that has been employed with modest success in multiple prospective trials.
3. Which patients are good candidates for treatment with immunosuppressive therapy (IST)?
In the phase III trial of antithymocyte globulin and cyclosporine versus best supportive care in patients primarily with lower-risk MDS, IST was shown to increase hematologic response rates significantly by 6 months (29% vs. 9%, respectively), but neither 2-year progression-free survival (46% vs. 55%) nor overall survival (49% vs. 63%) was significantly improved. Based on their previous experiences with IST, the National Institutes of Health (NIH) developed and validated a predictive score for response based on age, the presence of a human leukocyte antigen (HLA)-DR15 class II phenotype, and the duration of RBC transfusion dependence. The patient’s age in years is added to the duration of RBC transfusion dependence in months. A sum ≤57 predicts a high probability of response for patients in whom HLA-DR15 is absent, while a sum of ≤71 predicts higher responses for patients in whom HLA-DR15 is present. In addition to the above factors, other studies have reported that bone marrow hypocellularity, the presence of a paroxysmal nocturnal hemoglobinuria clone, and a low CD4:CD8 ratio correlate with improved response rates. We wait 4–6 months after therapy to assess response, and we closely observe for treatment-related toxicity such as infections and cyclosporine toxicity.
4. What is the best starting dose for lenalidomide in lower-risk patients with RBC transfusion dependence and a chromosome 5q deletion [del(5q)]?
The US Food and Drug Administration (FDA)-approved starting dose for lenalidomide in MDS is 10 mg daily continuously or for 21 days every 4 weeks based on the MDS-003 phase II registration trial conducted exclusively in transfusion-dependent patients with del(5q). A subsequent phase III trial randomized patients to placebo or two different doses of lenalidomide: 5 mg daily on days 1–28 and 10 mg daily on days 1–21 of a 28-day cycle. This trial was not designed to detect differences in the two lenalidomide arms, but higher rates of transfusion independence and cytogenetic response were seen in the 10 mg arm. In the phase II MDS-003 trial in patients with del(5q), patients were also originally treated with 10 mg on days 1–21, but shortly after activation, the schedule was amended to 10 mg continuous daily dosing in light of the faster times to response observed in the pilot trial. Forty-six patients received the day 1–21 schedule, and 102 received continuous daily dosing, which resulted in a slight trend toward higher erythroid response rates (72% vs. 77%, respectively; P = 0.26) and faster median time to response (4.7 weeks vs. 4.3 weeks). The 10 mg continuous dosing is associated with more frequent neutropenia and thrombocytopenia, but the development of these events is also associated with a higher probability of transfusion independence. Based on the available data, we initiate patients on the 10 mg continuous daily dosing schedule with weekly blood counts for the first 8 weeks. Approximately 80% of patients will require a drug holiday after a median duration of 3 weeks for myelosuppression. Treatment is held for an average of 3 weeks, then the dose is reduced to 5 mg daily. G-CSF can be used to accelerate neutrophil recovery and preempt neutropenia. The median time to response is 4 weeks, and the median duration of response is approximately 3 years.
5. Can lenalidomide be used in lower-risk patients with anemia and karyotypes other than del(5q)?
The use of lenalidomide in MDS without the del(5q) abnormality is considered off-label; however, a multicenter phase II trial (MDS-002) demonstrated some activity of this agent in lower-risk, transfusion-dependent MDS patients without this specific cytogenetic abnormality. In that trial, 26% of patients achieved RBC transfusion independence that lasted a median of 41 weeks. An additional 17% of patients had at least a 50% reduction in transfusion requirement, for an overall response rate of 43%. The median time to response (4.8 weeks) in this trial appears similar to the results in patients with del(5q). Unlike the MDS-003 study, however, there were very few cytogenetic responses. Further comparisons between the MDS-002 and MDS-003 trials show a shorter median duration of transfusion independence and a less robust median increase in hemoglobin in patients without the del(5q) abnormality. The exact role of lenalidomide in this specific population of patients is currently under investigation in a phase III study. Until the results of this trial are available, we consider this as a treatment alternative in lower-risk patients requiring frequent transfusions of RBCs who have failed ESAs, have adequate platelet and neutrophil counts, and otherwise do not have favorable characteristics for immunosuppressive therapy. The National Comprehensive Cancer Center Network (NCCN) guidelines for management of MDS list lenalidomide as an option for treatment of anemia in non-del(5q) lower-risk MDS.
6. Can patients with higher-risk disease and del(5q) be treated with lenalidomide rather than hypomethylating agents (HMAs)?
Lenalidomide treatment of higher-risk MDS with del(5q) should be considered investigational, and in our practice we use lenalidomide exclusively in lower-risk patients. A phase II study of daily lenalidomide in 47 higher-risk MDS patients with del(5q) reported a 27% response rate, including seven complete remissions (CRs). Most responses were rapid, but the duration of response was only 6.5 months. Patients with an isolated del(5q) were more likely to respond compared with those with additional chromosomal abnormalities. While this study utilized lenalidomide at the currently approved dose of 10 mg daily, dose escalation may improve response rates.
7. Does lenalidomide increase the risk of progression to acute myeloid leukemia (AML)? Should patients be monitored for clonal evolution while on lenalidomide therapy?
As seen in the long-term follow-up of 42 European patients treated in the MDS-003 trial, 15 patients (36%) progressed to AML and 17 (40%) had karyotypic evolution. Patients who failed to achieve a response to lenalidomide appeared to have a higher rate of AML progression compared to those who responded. These rates of AML progression in this isolated subset of patients may seem higher than historical controls, but patients in the MDS-003 trial were all transfusion dependent, which is a known poor prognostic factor for disease progression. In addition, recent studies indicate that TP53 gene mutations are demonstrable in approximately 20% of del(5q) MDS patients, expand over time, and are associated with higher risk of disease progression and lower frequency of cytogenetic response to lenalidomide. The rate of AML progression can be as high as 80% in patients with del(5q) and more than 5% blasts; thus, the apparent tendency toward leukemic transformation may actually be a reflection of the natural history of disease in this subset of patients with greater AML potential. A more recent study from the International Working Group on MDS with del(5q) evaluated 295 lenalidomide-treated patients on the MDS-003 and -004 studies along with 125 untreated, lower-risk, RBC-transfusion-dependent patients with del(5q) from a registry. The median follow-up was over 4 years in each cohort. Despite a higher RBC transfusion burden in the lenalidomide cohort, the 2-year cumulative AML progression risk was similar between cohorts [hazard ratio (HR) 0.969], but lenalidomide-treated patients had a significant improvement in survival (HR 0.597). In the phase III trial of lenalidomide versus placebo in del(5q) MDS, almost all patients who were randomized to placebo crossed over to the lenalidomide arm; thus, truly randomized prospective data with long-term follow-up are lacking, and the issue of whether lenalidomide truly increases the risk of AML progression remains unclear. Nonetheless, responding patients have a lower frequency of AML than nonresponders, suggesting that a drug effect is unlikely. No formal guidelines currently exist for monitoring cytogenetics in patients on lenalidomide, and we do not routinely perform karyotyping or fluorescent in situ hybridization on our responding patients.
8. How does one manage MDS patients with isolated thrombocytopenia? Are romiplostim or eltrombopag reasonable options?
Related posts:
 17: Management of Therapy-Related Myeloid Neoplasms
28: Chronic Myelomonocytic Leukemia
59: Autologous Hematopoietic Cell Transplantation in Multiple Myeloma
88: Recurrent and Metastatic Colorectal Cancer: Controversies, Consensus, and New Targets
17: Management of Therapy-Related Myeloid Neoplasms
28: Chronic Myelomonocytic Leukemia
59: Autologous Hematopoietic Cell Transplantation in Multiple Myeloma
88: Recurrent and Metastatic Colorectal Cancer: Controversies, Consensus, and New Targets
 11: Minimal Residual Disease in Acute Myeloid Leukemia
11: Minimal Residual Disease in Acute Myeloid Leukemia
 71: Secondary Brain and Spinal Cord Tumors
71: Secondary Brain and Spinal Cord Tumors



