© Springer International Publishing AG 2017
Doruk Erkan and Michael D. Lockshin (eds.)Antiphospholipid Syndrome10.1007/978-3-319-55442-6_1818. 15th International Congress on Antiphospholipid Antibodies Task Force on Antiphospholipid Syndrome Treatment Trends Report
Danieli Andrade1 , Ricard Cervera10, Hannah Cohen11, Mark Crowther12, Maria J. Cuadrado13, Guillaume Canaud14, David A. Garcia15, Maria Gerosa16, Thomas L. Ortel17, Vittorio Pengo2, Anisur Rahman3, Jane E. Salmon4, Rohan Willis5, Scott C. Woller6, 7, Doruk Erkan8, Michael D. Lockshin8 and Maria G. Tektonidou9
(1)
Department of Rheumatology, Hospital Das Clinicas, Sao Paulo, Brazil
(2)
Cardiac Thoracic and Vascular Sciences, University Hospital, Padova, Italy
(3)
Centre for Rheumatology Research, Division of Medicine, University College London Hospitals, London, UK
(4)
Department of Medicine, Hospital for Special Surgery – Weill Cornell Medicine, New York, NY, USA
(5)
Internal Medicine, Rheumatology Division, University of Texas Medical Branch, Galveston, TX, USA
(6)
Thrombosis Program, Intermountain Medical Center, Murray, UT, USA
(7)
University of Utah School of Medicine, Salt Lake City, UT, USA
(8)
Department of Rheumatology, Hospital for Special Surgery, Weill Cornell Medicine, New York, NY, USA
(9)
First Department of Propaedeutic Internal Medicine, Laiko Hospital, Athens, Greece
(10)
Department of Autoimmune Diseases, Hospital Clínic, Barcelona, Catalonia, Spain
(11)
Haemostasis Research Unit, Department of Haematology, University College London, London, UK
(12)
Pathology and Molecular Medicine, St Joseph’s Hospital and McMaster University, Hamilton, ON, Canada
(13)
Rheumatology/Lupus Unit, Guys and St Thomas NHS Foundation Trust, London, UK
(14)
Renal Division, Necker Hospital & Paris Descartes University, Paris, France
(15)
Department of Medicine, Division of Hematology, University of Washington Medical Center, Seattle, WA, USA
(16)
Division of Rheumatology, Gaetano Pini-CTO Hospital, Clinical Science and Community Health, University of Milan, Milan, Italy
(17)
Department of Medicine, Duke University Medical Center, Durham, NC, USA
Keywords
Direct Oral AnticoagulantsStatinsComplement InhibitionmTOR InhibitionVitamin DAnti Peptide TherapyB cell InhibitionClopidogrelIntegrin inhibitorsDefibrotideCilostazolProtease-activator Receptor AntagonistsToll-like Receptor InhibitorsTissue Factor InhibitorsIntroduction
Antiphospholipid syndrome (APS) is characterized by arterial and/or venous thrombosis , pregnancy morbidity, and persistent antiphospholipid antibodies [1]. Thrombosis and pregnancy morbidity (recurrent embryonic or fetal loss, preeclampsia [PE], and intrauterine growth restriction [IUGR]) compose the criteria manifestations. Non-criteria manifestations (livedo reticularis, thrombocytopenia, hemolytic anemia, cardiac valve disease, nephropathy, skin ulcers, and cognitive dysfunction) are also part of the disease spectrum. Catastrophic antiphospholipid syndrome (CAPS) is a rare but severe disease manifestation associated with high mortality.
Anticoagulation with vitamin K antagonists (VKA) is conventional therapy for secondary thrombosis prevention, but strict laboratory monitoring, dietary modifications, and medication adherence are required for optimal treatment. Despite anticoagulation, thrombosis recurrence can be as high as 40% after 10-year follow-up [2]. Non-criteria manifestations are usually refractory to anticoagulation; their management is based on anecdotal experience.
The goal of the 14th International Congress on Antiphospholipid Antibodies (Rio De Janeiro, Brazil, September 2013) Task Force on APS Treatment Trends was to offer opinions on potential new treatment strategies, other than conventional anticoagulants or antiplatelet agents. The task force members systematically reviewed in vitro, animal, and completed or ongoing clinical studies in aPL-positive patients. Recommendations were presented in open discussions before and during the congress, following which the task force report was finalized and published [3].
The goal of the 15th International Congress on Antiphospholipid Antibodies (www.apsistanbul2016.org) (North Cyprus, September 2016) Task Force on APS Treatment Trends was to update potential future treatments. The task force members again systematically reviewed the most recent literature, presented recommendations during the Congress, and finalized the report. It is organized in two parts: (a) update on the treatments included in the first report (please refer to the original report for detailed information) [3] and (b) new treatments and/or pathways for consideration.
Part A: Update on Treatments Discussed Previously
Direct Oral Anticoagulants
Direct oral anticoagulants (DOACs) include the direct thrombin inhibitor dabigatran etexilate and the direct anti-factor Xa inhibitors rivaroxaban, apixaban, and edoxaban. These agents, unlike warfarin, are prescribed in fixed doses with more predictable anticoagulant effects, do not interact with diet or alcohol, and have fewer reported drug interactions that affect anticoagulant intensity. Furthermore, monitoring of anticoagulant intensity of a DOAC is not routinely required because anticoagulant effects are more predictable.
The 14th International Congress on aPL Task Force on Treatment Trends concluded that “warfarin or other VKA remains the mainstay of anticoagulation in thrombotic APS. Direct oral antiocagulants can be considered in APS patients with a first or recurrent venous thromboembolism (VTE) occurring off or on subtherapeutic anticoagulation, only when there is known VKA allergy/intolerance or poor anticoagulant control. There were no data to recommend DOACs in APS patients with recurrent VTE occurring on therapeutic anticoagulation or with APS-related arterial thrombosis” [3].
A systematic review (through April 2016) presented during the 15th International Congress on aPL identified seven case reports and four case series . They included 99 DOAC-treated APS patients, of whom 38 had primary APS, 23 APS associated with lupus, and 38 unspecified. Approximately 20% of patients had vascular events during a mean follow-up of 12 months [4]. More recently, in a prospective case series, six (four triple aPL-positive) of 56 (11%) APS patients developed recurrent thrombosis on DOAC (mean follow-up 22 months) [5].
The anecdotal clinical reports (in which rivaroxaban was used in the majority of patients), with recognition of their inherent limitations of publication bias and low evidence level study designs, suggest that recurrent thrombotic events with DOACs in APS patients mainly occur when DOACs are used for secondary prevention of aPL-related arterial thrombosis or microthrombosis, situations where DOACs are not approved. They highlight the need for randomized controlled trials to guide the use of DOACs in thrombotic APS. One recently completed and two ongoing controlled clinical trials and a feasibility study on DOACS were presented during the Congress.
Rivaroxaban in Antiphospholipid Syndrome (RAPS) Trial
The Rivaroxaban in Antiphospholipid Syndrome (RAPS) trial (principal investigator [PI], Hannah Cohen) compared rivaroxaban to warfarin (at a target international normalized ratio [INR] of 2.5, range 2.0–3.0) to treat patients with previous VTE; RAPS was a randomized, controlled, open-label, phase II/III, non-inferiority trial [6]. Eligible patients had taken standard-intensity warfarin for at least 3 months after the last venous thromboembolism (VTE). Exclusion criteria were arterial thrombosis, recurrent VTE while taking warfarin at a therapeutic INR, and age younger than 18 years. Warfarin-treated APS patients with previous VTE, with or without SLE, were randomized 1:1 to warfarin or rivaroxaban, 20 mg once daily, stratified by center and SLE/non-SLE.
The definition of non-inferiority was not VTE recurrence rate but thrombin generation testing, which assesses in vitro the inhibitory effects of anticoagulants. The primary outcome measure was percentage change in endogenous thrombin potential (ETP, the area under the thrombin generation curve) from randomization to day 42, with treatment continued for 180 days and follow-up for 210 days.
One hundred sixteen patients were randomized. Judged by the primary outcome, percentage change in ETP, rivaroxaban was inferior to warfarin . However, because peak thrombin generation was lower with rivaroxaban, the overall thrombogram suggested no difference in thrombotic risk. No new thrombotic events were seen during 6 months of treatment. No patients had major bleeding; clinically relevant and minor bleeding rates were similar in the two groups.
A limitation of the RAPS is that it used a laboratory surrogate outcome measure. The intended selection bias, limiting the selection of patients to those with previous VTE leading to treatment with standard-intensity warfarin, and which ensured a clinically homogeneous study population with definite APS, was a strength. The authors cautioned that the results do not apply to APS patients with venous thrombosis who require higher intensity anticoagulation and to APS patients with arterial thrombosis.
Rivaroxaban in Thrombotic Antiphospholipid Syndrome (TRAPS) Trial
The objective of the ongoing TRAPS trial (PI, Vittorio Pengo) is to demonstrate non-inferiority of rivaroxaban 20 mg (15 mg in patients with moderate renal insufficiency) daily versus warfarin (INR 2.0–3.0) with respect to cumulative incident thrombosis (arterial or venous) confirmed by imaging studies, major bleed, and death in triple aPL-positive APS patients. The trial is multicenter, interventional, prospective, parallel, randomized, controlled, and open-label trial; it plans to recruit 535 patients [7].
Rivaroxaban in Antiphospholipid Syndrome (RAPS ) Pilot Feasibility Study
The rivaroxaban in APS pilot feasibility study (PI, Mark Crowther) (clinicaltrials.gov#: NCT02116036) is a prospective cohort study for patients with confirmed APS and prior VTE, with or without prior arterial thrombosis, allocating them to receive rivaroxaban, 20 mg daily. Patients are followed for thrombosis. The study is designed as a feasibility study with clinical outcomes as secondary endpoints (thrombosis; minor, major, and fatal bleeding). Recruitment was closed on September 30, 2016, with a plan to follow all patients for 1 year. Seventy-nine patients were identified, far below the expected recruitment of 150, suggesting that future studies will have to employ many centers and be international. To date few complications, and no recurrent thromboses, have occurred. One patient suffered unexplained hepatitis. This Canadian RAPS study will be underpowered but will add to the body of evidence on safety and efficacy of DOACs in patients with APS.
Apixaban for the Secondary Thrombosis Prevention in Antiphospholipid Syndrome (ASTRO-APS )
ASTRO-APS (PI, Scott Woller) is a prospective, randomized, open-label, blinded pilot study comparing apixaban with dose-adjusted warfarin (target INR range 2.0–3.0) for the secondary prevention of thromboembolism among patients with a history of APS and thrombosis. The intentions of this phase IV pilot study are to provide data on feasibility of enrolling APS patients and to estimate efficacy and safety of apixaban compared with usual care [8].
ASTRO-APS was originally designed to compare apixaban 2.5 mg twice a day with dose-adjusted warfarin, enrolling patients with history of arterial or venous thromboses receiving indefinite anticoagulation. The primary clinical outcomes are rates at 1 year of arterial or venous thrombosis, death caused by thrombosis, major bleeding, and clinically relevant nonmajor bleeding.
After accrual of the first 25 patients, a prespecified Data Safety Monitoring Board (DSMB) review recommended the protocol be modified to use apixaban 5 mg twice a day. After five more patients were enrolled, a potential safety signal led to an ad hoc DSMB rereview, which recommended: first, to continue ASTRO-APS; second, exclude patients with prior arterial thrombosis; and, third, obtain brain magnetic resonance imaging with stroke protocol for all otherwise eligible candidates to exclude prior silent stroke. ASTRO-APS plans to enroll 200 patients.
15th International Congress on Antiphospholipid Antibodies Task Force on Treatment Trends Recommendation
Insufficient evidence exists to make recommendations at this time regarding DOAC use in APS. The RAPS trial suggests that rivaroxaban might be useful in selected APS patients with single venous thromboembolism requiring standard-intensity anticoagulation; however, this needs to be confirmed with additional studies using clinical outcome measures.
Statins
Benefit of statins in primary and secondary prevention of coronary heart disease is proven, which is due to the lipid-lowering effect of these drugs and to their immunomodulatory, anti-inflammatory, and antithrombotic properties [9]. Statins have multiple effects on monocyte, lymphocyte, and endothelial cell activities that may contribute to thrombosis prevention in APS. Antiphospholipid antibodies induce expression of tissue factor (TF) and cell adhesion molecules; fluvastatin, simvastatin, and rosuvastatin reduce this expression [10]. In a mouse model of obstetrical APS, simvastatin and pravastatin reduced fetal death by inhibiting TF and protease-activated receptor 2 (PAR2) expression on neutrophils [11]. In a thrombosis mouse model, fluvastatin reduced thrombus size [12].
Based on in vitro and two human studies using surrogate markers [13, 14], the 14th International Congress on aPL Task Force on Treatment Trends concluded that “although statins ameliorate the proinflammatory profile and down-regulate the prothrombotic stage found in APS patients, based on the available data, statins cannot be recommended in APS patients in the absence of hyperlipidemia. However, a subgroup of aPL-positive patients with recurrent thrombosis despite adequate anticoagulation might derive benefits from statins [3].”
Since then there have been no systematic studies with statins in thrombotic APS. A study presented at the 15th International Congress on aPL [15] reported 21 pregnant APS patients who developed preeclampsia (PE) and/or IUGR on low-dose aspirin (LDA) and low molecular weight heparin (LMWH). Of those studied, 11 received pravastatin (20 mg daily) initiated at the onset of PE and/or IUGR (in addition to LDA + LMWH) and 10 did not. The study was not randomized, and not all patients met APS criteria at the time of enrollment. All pravastatin-treated patients had live births near full term. In the 11 patients who did not receive pravastatin, 11 deliveries were preterm, and five neonates died, and three of the six survivors had abnormal development. Patients treated with pravastatin had increased placental blood flow and improvements in PE features as early as 10 days after treatment was begun.
The rapid improvement in uterine artery hemodynamic parameters in pravastatin-treated patients (uteroplacental perfusion was assessed by Doppler) suggests that the drug targets placental vasculopathy, possibly by stimulating release of vasoactive substances, such as nitric oxide, from the endothelium [16]. Statins also downregulate TF, the major initiator of the coagulation cascade in vivo and a crucial molecule linking inflammation and thrombosis in APS [17]. These protective effects may explain the amelioration of placental and maternal PE [18].
15th International Congress on Antiphospholipid Antibodies Task Force on Treatment Trends Recommendation
No controlled clinical trial supports the use of statins in APS patients. Because statins downregulate prothrombotic and proinflammatory biomarkers, statins may be used in APS with high risk for cardiovascular events and in those with recurrent thrombosis despite adequate anticoagulation.
Statins are classified as Category X (contraindicated) for pregnancy by the US Food and Drug Administration, because of the disruption of gonadal stem cell development and theoretical long-term fetal neurological damage [19]; however, several recent studies did not find teratogenic effects [20, 21]. An American national study on drugs in pregnancy [21] did not find an increase in congenital malformations or organ-specific malformations among the offspring of the 1152 women exposed to statins during the first trimester; the relative risk of malformation was 1.79 (95% confidence interval 1.43–2.23), which fell to 1.07 when controlled for the confounders such as diabetes. The task force does not suggest the use of statins during APS pregnancies without further efficacy and safety data.
B-Cell Inhibition
Animal models demonstrate that B cells play an important role in the pathogenesis of APS. Blocking B-cell-activating factor (BAFF) prevents disease onset and prolongs survival in APS murine models [23]. Rituximab and belimumab are the only B-cell-inhibiting biologic therapies whose effect has been studied in APS patients.
Based on the case reports [23–25], CAPS registry data [26], and one open-label uncontrolled pilot study [27], the 14th International Congress on aPL Task Force on Treatment Trends concluded that “B-cell inhibition may have a role in difficult-to-treat APS patients, possibly in those with hematologic and microthrombotic/microangiopathic manifestations [3].”
Since then, although there have been case reports/series of rituximab use in APS and a recent report of belimumab use in two primary APS patients [28], there is not yet a prospective study of B-cell inhibition in APS.
15th International Congress on Antiphospholipid Antibodies Task Force on Treatment Trends Recommendation
No change in recommendations.
Complement Inhibition
Complement activation contributes to thrombosis in APS animal models. Antiphospholipid antibodies activate complement factor 5 (C5), generating fragment 5a (C5a), which induces adhesion molecules and TF in inflammatory cells and platelets and triggers release of prothrombotic and pro-inflammatory mediators [29, 30]. In animal models of APS, C5a interacts with its receptor (C5aR), amplifying endothelial cell activation and vascular inflammation, promoting trophoblast injury and angiogenic factor imbalance, and producing microthrombotic lesions [29, 31, 32]. Inhibition by anti-C5 antibody, C5aR antagonist peptides, and anti-C5aR antibody protects mice from pregnancy losses and from thrombosis [33]. Mice treated with aPL but deficient in complement regulatory components, i.e., mice in which the complement cascade is not normally inhibited, have poor pregnancy outcomes, including thrombotic microangiopathy. Mice treated with aPL, but deficient in complement components, have good pregnancy outcomes. These contrasting experiments indicate that complement is an important mediator in pathogenesis of both manifestations of APS [34]. Heparin, used in APS patients to treat thrombosis and prevent miscarriages, has a complement inhibitory effect [35].
Given the reports that complement inhibition improves outcomes in mouse models and CAPS patients [36], the 14th International Congress on aPL Task Force on Treatment Trends concluded that “complement inhibition may have a role as an adjuvant or main therapy for APS patients refractory to anticoagulation; however more clinical data are needed before this medication can be recommended” [3].
Since then a phase IIa study of treatment of non-criteria manifestations of APS (nephropathy, thrombocytopenia, and skin ulcers), designed to evaluate safety of an intravenous C5a inhibitor (clinicaltrials.gov #: NCT02128269), has ended, but the results are not yet available. Another phase II study using eculizumab (a C5 inhibitor) to prevent thrombosis after renal transplantation in patients with prior CAPS (clinicaltrials.gov#: NCT1029587) is currently recruiting.
Eculizumab is a fully recombinant humanized hybrid IgG2/IgG4 monoclonal antibody that binds to C5 and inhibits C5 cleavage by C5 convertase, thereby inhibiting the inflammatory, thrombotic, and lytic functions of complement. Eculizumab is FDA-approved for paroxysmal nocturnal hemoglobinuria (PNH) , an illness in which red blood cells and leukocytes are prone to complement-mediated lysis due to deficiency in CD55 or CD59, and for atypical hemolytic-uremic syndrome (HUS) [37]. A case report described complement inhibition in a refractory patient with CAPS who underwent kidney transplantation [38], and several other cases have been published [36]. Kidney allograft survival improves if complement blockade is initiated at the time of transplantation [39], suggesting a use in patients with APS nephropathy undergoing renal transplantation.
Complement plays a role in ischemia/reperfusion injury, antibody- and cell-mediated transplant rejection, C3 glomerulopathy, and atypical HUS [40]. Eculizumab was used to block thrombotic microangiopathy (TMA) in a patient with atypical HUS [41]. Three patients with APS (two with CAPS) treated with anticoagulation and eculizumab prior to and after renal transplantation had functioning renal allografts, with no systemic thrombotic events or early graft losses [42], confirmed in a report demonstrating improvement in TMA after eculizumab in APS nephropathy that recurred after kidney transplantation. In this case pretreatment intense C5b-9 and C4d deposit in kidney biopsies were absent after treatment with eculizumab, but chronic vascular renal changes were not prevented [43]. A recent case report described the potential use of eculizumab to prevent re-thrombosis in an arterial bypass graft in APS. Immunofluorescence showing β2-glycoprotein-I (β2GPI) on the endothelium of the artery wall suggested a pathogenic role for aPL [44].
Rivaroxaban, a direct factor Xa inhibitor, decreases markers of complement activation (C3a, C5a, and soluble [s] C5b-9) [45]. Antiphospholipid syndrome patients had higher baseline C3a, C5a, and SC5b-9 and, after 42 days of rivaroxaban treatment, a statistically significant decrease was observed (compared to controls treated with warfarin), possibly because rivaroxaban inhibits FXa-induced complement activation of C5 [46]. Further studies are necessary to confirm these findings.
Recently a novel recombinant antibody recognizing the first domain of β2GPI was shown to induce thrombosis and fetal loss in animal models [47]; a non-complement fixing CH2-deleted variant of that antibody reduced the detrimental effects, suggesting another complement-mediated mechanism and another possible treatment for refractory APS [47].
15th International Congress on Antiphospholipid Antibodies Task Force on Treatment Trends Recommendation
Although there are encouraging reports about efficacy of complement inhibition in APS, publications may be biased toward positive results. At this time, patients with life-threatening disease resistant to other interventions may be candidates for complement inhibitors as salvage therapy.
Peptide Therapy
All proposed peptide therapies are based on the premise that the key pathogenic interaction in patients with APS occurs when aPL engages one of the five domains (DI–DV) of β2GPI [48]. Antibodies to several domains occur in APS patients, but most pathogenic aPL bind the N-terminal DI [49]. Each antigen-binding arm of the antibody can bind a separate β2GPI molecule, creating a dimeric structure that binds anionic PL on cell membranes, thus interacting with cell surface receptors like Toll-like receptors (TLR) and apolipoprotein E receptor 2 (ApoER2) to alter cell function [50]. In the absence of aPL, β2GPI is monomeric, is present constitutively in human serum, and does not exert these effects. The peptide agents proposed as new therapies for APS all act by blocking binding at different points. It is unknown which approach is best.
The 14th International Congress on aPL Task Force on Treatment Trends task force recommended that “at present, peptide therapy is not ready for trials in patients; however peptide therapy is potentially an important future targeted treatment for aPL-positive patients. Chemical modification to improve half-life and minimize immunogenicity will be required. Different peptides may be needed for different aPL manifestations” [3].
Since then, no peptide therapies have yet entered human trials. Considerable information from in vitro and animal studies suggests that peptides may become therapeutic agents in the future.
It is unknown which domain of β2GPI may be the best therapeutic target. The key epitope for aPL on DI lies between the arginine residues R39 and R43 [49, 51]. In the in vivo femoral vein thrombosis model developed by Silvia Pierangeli, some DI peptide analogues inhibited thrombosis, TF expression in peritoneal macrophages, and aortic VCAM-1 expression, whereas others did not [52].
Linear DI peptides containing the critical R39–R43 epitope do not bind APS-IgG as well as does whole DI [53, 54]. McDonnell et al. have published a method for producing recombinant DI in bacteria [55] and have improved its half-life by adding polyethylene glycol group PEGylation [56, 57].
An alternative idea is to block DV binding to phospholipids. The octapeptide CKNKEKKC inhibits binding aPL to cardiolipin [58]. A 15-mer peptide from DV (called GDKV) and a cytomegalovirus peptide (TIFI) homologous to GDKV have been used as inhibitors. In mouse models that use injected aPL, TIFI inhibits thrombosis in the femoral vein [59] and reduces fetal loss [60]. TIFI also reduces binding of β2GPI to human umbilical vein endothelial cells (HUVEC) [59] and to human trophoblast cells [60].
Blank et al. joined two synthetic peptides derived from DV (one was GDKV) with a flexible linker molecule [61]. When HUVEC were preincubated for a short while with the construct, binding of β2GPI/anti-β2GPI antibody (aβ2GPI) complexes to HUVEC was reduced by 89%, but the reduction in binding was lost with prolonged preincubation because the construct was taken up in the cells, thus tempering enthusiasm for use of this agent.
Kolyada et al. developed a dimer, the A1 ligand-binding module of the ApoER2 receptor, that binds DV of β2GPI [62–64]. Their construct blocks binding of both β2GPI/ aβ2GPI and DV alone to cardiolipin far more strongly than does monomeric A1 but does not do so in the absence of aβ2GPI; it reduces thrombosis induced by laser trauma in two mouse models [63]. Recently the same group developed a mutant dimer that inhibits binding and clotting more strongly than does the wild type [64].
Recombinant DI, TIFI, and A1-A1 are all credible peptide therapies for APS. Proponents of TIFI and A1-A1 could argue that the heterogeneity of the aβ2-glycoprotein-I in patients with APS mitigates against an approach that targets DI alone. Conversely, others might argue that interfering with the interaction of DV with PL may have adverse effects on physiological processes that occur in the absence of aPL. Perhaps there is a need for different peptide therapies. There is little information about potential toxic effects of any of these agents. All putative peptide therapies will have to be modified to enhance half-life [63].
15th International Congress on Antiphospholipid Antibodies Task Force on Treatment Trends Recommendation
The key research needs are to take one or more of these agents forward to formal pharmacokinetic and toxicology studies, then to a first-in-man trial.
Vitamin D
A growing body of evidence highlights vitamin D’s immunomodulatory properties. Vitamin D insufficiency (<30 ng/ml) occurs in up to 70% of patients with APS and/or SLE, and vitamin D deficiency (<10 ng/ml) occurs in 11–50%. Low vitamin D levels in APS patients correlate with venous and arterial thrombosis and with non-criteria manifestations. Values in thrombotic APS patients are lower than in obstetric APS patients [65–67]. However, low-dose short-term vitamin D supplementation in a small group of primary APS patients was ineffective in raising levels above 30 ng/ml [67]. Human umbilical vein endothelial cell cultures demonstrate that vitamin D inhibits expression of prothrombotic TF in response to aβ2GPI stimulation [65], a possible mechanism for therapeutic effect.
The 14th International Congress on aPL Task Force on Treatment Trends task force recommended that “vitamin D deficiency and insufficiency should be corrected in all aPL-positive patients based on the general population guidelines. The prognostic role of vitamin D deficiency and therapeutic value of supplementation (including the dosage and definition of treatment goals) in aPL-positive patients should be clarified with prospective studies that include appropriate control groups and standardized definitions of vitamin D deficiency” [3].
There have been no ongoing studies evaluating the effect of vitamin D in aPL-positive patients. However, a randomized clinical trial of vitamin D prophylaxis in the prevention of hypertensive disorders of pregnancy (clinicaltrials.gov #: NCT02920593) recently began. Investigators will also evaluate placental pathology and measure placental levels of several proinflammatory markers.
In the early stages of pregnancy, trophoblasts respond to and produce vitamin D, promoting an anti-inflammatory environment and inducing decidualization [68–70]. Vitamin D deficiency is associated with an increased risk for preeclampsia and IUGR [71]. A retrospective cross-sectional study of women with recurrent pregnancy loss (RPL) stated that vitamin D deficiency and insufficiency are associated with aPL, elevated peripheral blood natural killer (NK) cells, and elevated NK cell cytotoxicity [72]. In vitro studies confirmed the ameliorative effect of vitamin D on NK cell cytotoxicity and Th2-type response, both associated with successful pregnancy outcomes [72, 73].
Studies utilizing a human first trimester trophoblast cell line and primary trophoblast cultures show that vitamin D treatment alone or combined with LMWH limits aPL-mediated inflammatory response [74]. Vitamin D also reduces the elevated anti-angiogenic factor sFlt-1 seen in aPL- and/or LMWH-treated trophoblasts and placental villi explants [74–76]. The importance of this finding lies in the strong association of sFlt-1 with preeclampsia. Elevated levels of sFlt-1 are also seen in pregnant women treated with LMWH [77–78], possibly explaining the contradictory results of studies on the effectiveness of LMWH and aspirin in aPL-associated pregnancies [79] and suggesting a role for adjunctive vitamin D in LMWH-treated obstetric APS.
Related posts:
 Recent Advances in Understanding of the Genetics of Antiphospholipid Syndrome
Clinical and Prognostic Significance of Non-criteria Antiphospholipid Antibody Tests
Prevention and Treatment of Obstetric Antiphospholipid Syndrome
15th International Congress on Antiphospholipid Antibodies Task Force on Catastrophic Antiphospholipid Syndrome Report
Recent Advances in Understanding of the Genetics of Antiphospholipid Syndrome
Clinical and Prognostic Significance of Non-criteria Antiphospholipid Antibody Tests
Prevention and Treatment of Obstetric Antiphospholipid Syndrome
15th International Congress on Antiphospholipid Antibodies Task Force on Catastrophic Antiphospholipid Syndrome Report
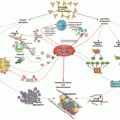 Mechanisms of Antiphospholipid Antibody-Mediated Thrombosis
Mechanisms of Antiphospholipid Antibody-Mediated Thrombosis
 Treatment of Non-criteria Manifestations in Antiphospholipid Syndrome
Treatment of Non-criteria Manifestations in Antiphospholipid Syndrome
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


