Epidemiology
ASM deficiency is a rare genetic disorder, and limited information is available regarding its demographics and incidence. Several studies have estimated the birth rate of this disorder at ~0.5 to 1 per 100,000 [1, 2]. However, these estimates relied on data from biochemical testing laboratories where suspected cases were referred by clinicians for enzymatic confirmation, rather than on population screening. Thus, they are only approximations, and probably under-represent the true disease frequency. This is particularly true for the late-onset (type B) cases, where the clinical diagnosis may not be readily apparent.
The only population where DNA-based screening has taken place for ASM-deficient NPD is the Ashkenazi Jewish community, where the carrier frequency of three common mutations causing the type A form of the disorder has been estimated to be between ~1:90 and 1:120 [3, 4]. This suggests an estimated birth rate of ~2–3 per 100,000 for this form of the disorder among Ashkenazi Jewish individuals.
Demographic data is currently available at the Mount Sinai School of Medicine for ~1,000 individuals referred for mutational analysis over a 20-year period (Schuchman et al., unpublished information). About 300 of these patients were referred with an infantile, neurological phenotype (“type A” NPD), and of these, ~60% were Ashkenazi Jewish. The remaining patients derived from over 40 countries, and in contrast to the type A patients, very few of these individuals had Ashkenazi Jewish ancestry. The majority of these individuals were from North America and western Europe, although this may be reflective of enhanced awareness and diagnosis of lysosomal storage disorders in these regions, rather than a higher disease frequency. A significant number also were from North Africa and the Middle East, suggesting that ASM deficiency may be more common in these regions than was previously thought. In fact, over 40% of the NPD patients referred to Mount Sinai from Europe were of North African, Turkish or Arab ancestry. A significant number also were referred from South America, particularly Chile and Brazil. Asia had the fewest number of ASM-deficient NPD cases referred to our centre.
Genetics
The SMPD1 gene is located on the short arm of chromosome 11 (11p15.4) [5]. The gene spans ~6 kb and consists of 6 exons encoding the 629 amino acids of the full-length ASM polypeptide [6]. Notably, SMPD1 is the only gene encoding a lysosomal protein for which genomic imprinting has been demonstrated; i.e. it is preferentially expressed from the maternal chromosome (paternally imprinted) [7].
The clinical presentation of ASM-deficient NPD is largely dependent on the type of mutation(s) the patient inherits, and the resultant effect(s) of these mutations on the residual ASM polypeptide. However, because the SMPD1 gene is imprinted, the phenotypic variability may also be due, at least in part, to the inheritance of specific mutations on the maternal vs. paternal alleles. In addition, abnormal clinical and laboratory findings have been reported in heterozygous individuals carrying only one SMPD1 mutation [8]. This could similarly be due to the inheritance of a single, “severe” SMPD1 mutation on the preferentially expressed maternal chromosome.
To date, over 100 mutations have been found within the SMPD1 gene causing ASM-deficient NPD (http://www.hgmd.org/). These include point mutations, small deletions, and splicing abnormalities. No “hot-spots” exist for these mutations, although since ~40% of the ASM polypeptide is encoded by exon 2 of the SMPD1 gene, a preponderance of mutations can be found within this region. Several polymorphisms also have been found within this gene, including a varying number of repeated nucleotides within the ASM signal peptide region [9, 10]. In addition, there are two in-frame ATG initiation codons within the SMPD1 gene, and mutation analysis has revealed that both may be functional [11]. Because the SMPD1 cDNA and gene may vary in length, the nomenclature for the same mutation may differ between reports based on the reference sequence that is used. For example, the p.deltaR608 mutation originally described by Levran et al [12] may also be referred to as p.deltaR610.
Identification of mutations in ASM-deficient NPD patients has permitted the first genotype/phenotype correlations for this disorder, and the first genetic screening efforts. Several mutations have been found within specific populations leading to severe, intermediate, or less severe phenotypes. For example, three mutations account for over 90% of Ashkenazi Jewish type A NPD infants [13–15], and homozygosity for these mutations (or the presence of one of these mutations in combination with another) is predictive of the severe NPD form. Another mutation, p.deltaR608, only occurs in non-neurological ASM-deficient NPD patients, and is found in ~15–20% of such individuals in western Europe and North America [12]. This mutation is considered “neuroprotective” since, even when found together with one of the severe type A mutations, the patients lack neurological involvement. The deltaR608 mutation is also prevalent in NPD patients from North Africa [16]. Yet another mutation, p.Q292K, is associated with an intermediate, neurological phenotype [17]. These and other findings have assisted physicians, genetic counselors and families in predicting the phenotypic outcome in individual ASM-deficient NPD patients, and in the future may lead to large-scale screening for this disorder in other populations.
Pathophysiology
The primary accumulating lipid in ASM-deficient NPD is sphingomyelin, a major structural component of all cell membranes. Thus, unlike several of the other sphingolipid storage disorders (e.g. Gaucher, Fabry, Tay–Sachs, etc.), the primary lipid affected in ASM-deficient NPD is an abundant component of all cells. Sphingomyelin accumulation in cells from ASM-deficient patients may first be detected within the lysosomal/endosomal compartments, but is also found in the plasma membrane and other cellular membranes. These membrane abnormalities, in turn, may result in many downstream cellular changes, including abnormalities in cell signalling pathways, receptor function, transport mechanisms, etc. Moreover, as in all lysosomal disorders, , many other lipid (and non-lipid) molecules may accumulate in ASM-deficient NPD cells, secondary to the primary metabolic abnormality, including cholesterol, ceramide, sphingosine and others, contributing to the cellular dysfunction. Indeed, the early detection of cholesterol accumulation in ASM-deficient cells led to the misclassification of types A and B NPD, and type C NPD, as allelic forms of the same disorder. It is now known that type C NPD is a distinct disorder due to mutations in one of two genes encoding proteins involved in cholesterol transport (see Chapter 11).
Many of the secondary lipids accumulating in ASM-deficient NPD cells have important biological effects on cell survival and proliferation, which likely contributes to the disease pathogenesis (for a review, see [18]). For example, ceramide and sphingosine are important cell-signalling lipids that are abnormally expressed following cellular stress or in response to many common disease pathologies (e.g. diabetes, fibrosis, sepsis, cancer, etc.). The activity of ASM is a major source of ceramide in cells, and although the primary “housekeeping” function of the enzyme appears to be within the lysosome, it is also subject to rapid and specific translocation to the cell surface when cells are stressed, and can be readily detected in the circulation. The pathogenic consequence of the deficiency of cell surface and secreted ASM in NPD patients remains unknown, as is the direct effect of the secondary lipid abnormalities on the disease pathology.
The primary organ systems affected in all ASM-deficient patients are the spleen, liver and lung. Lipid-filled foam cells can be readily detected in the parenchymal tissues of the spleen and liver, as well as in the pulmonary airways. The onset and severity of pulmonary disease in ASM-deficient NPD is highly variable, and is primarily due to the infiltration of inflammatory cells into the airways. In ASM-deficient mice (see below), the infiltration of inflammatory cells can be correlated with the elevated release of lung chemokines, including MIP1alpha [19]. Once present, these inflammatory cells can be very long-lived, and in the case of ASM deficiency also exhibit defective phagocytosis and other biological properties (Figure 10.1).
Foam cells (often referred to as “NPD” cells) are readily detected in the bone marrow of ASM-deficient patients, although the effect of this enzyme deficiency on bone and cartilage disease has not been studied in much detail. It is notable that many adult ASM-deficient NPD patients exhibit joint and bone pain, and there may be a higher incidence of fractures in these patients as well. In addition, there is a growing literature showing the importance of sphingolipid metabolism, and sphingomyelin/ceramide in particular, on normal cartilage and bone homeostasis.
Due to the cellular abnormalities in the liver and spleen, ASM-deficient NPD patients often present with abnormal haematological and plasma lipid findings. For example, low platelets are a common finding in the disease, as is the combination of very low HDL, high LDL and high triglycerides. The consequences of these lipid abnormalities on cardiac disease in ASM deficiency is not clearly understood, although there is evidence for early cardiac calcifications in some patients and early cardiovascular disease. Growth abnormalities also are frequently found in children with ASM deficiency, and may be due to abnormalities in the IGF-1 signalling pathway [20].
Finally, in patients with type A NPD, the brain is severely affected. Although little is known about brain pathology in ASM-deficient NPD patients, a series of recent studies in the ASM knockout mice (see below) revealed numerous abnormalities, including Purkinje cell death in the cerebellum, as well as abnormal synaptic vesicle release and uptake, calcium homeostasis, neuronal polarization, myelin production and immune responses (for a review, see [21]).
Clinical presentation
Type A Niemann–Pick disease
Most patients with type A NPD present with hepatosplenomegaly at a median age of 3 months. Early development and attainment of developmental milestones are typically normal. Absence of deep tendon reflexes and hypotonia are the most common, early abnormalities on neurologic examination. At approximately 9 months of age, development plateaus and then regresses. Retinal cherry-red spots are apparent by 12 months of age. Many patients develop significant gastroesophageal reflux, which makes feeding difficult. The liver and spleen continue to enlarge until they occupy most of the abdominal and pelvic cavity, and some patients develop significant ascites. Chest radiographs reveal diffuse interstitial disease. Laboratory abnormalities include elevated levels of SGOT, SGPT, total cholesterol and triglycerides, low HDL-C and platelets, and elevated chitotriosidase levels.
Type B Niemann–Pick disease
Type B NPD is clinically heterogenous, and the age of clinical onset, disease manifestations, and severity varies considerably. Hepatosplenomegaly is the most common initial presentation of type B NPD, often presenting in childhood. Pulmonary disease is apparent on chest radiograph and high resolution chest CT, and many patients have low FEV1, FVC and DLCO on pulmonary function testing consistent with restrictive lung disease. Although children with type B NPD typically have bone age delay, delayed onset of puberty and short stature, most patients eventually reach a normal adult height. Many patients have early satiety due to gastric compression by the enlarged spleen. Decreased bone mineral density and osteopenia occur in the majority of patients, and pathologic fractures are not uncommon (McGovern et al. in preparation). Bleeding episodes, especially recurrent epistaxis, are common in younger patients. Hepatic manifestations can include elevated transaminases, hepatic fibrosis, cirrhosis, and ascites. Most patients have an abnormally low concentration of HDL-cholesterol, high total and LDL-cholesterol, and high plasma triglycerides. They are also at risk to develop coronary artery disease [22]. Although ophthalmic examination in about 25% of all type B patients will reveal cherry-red spots, most do not have abnormal neurological symptoms.
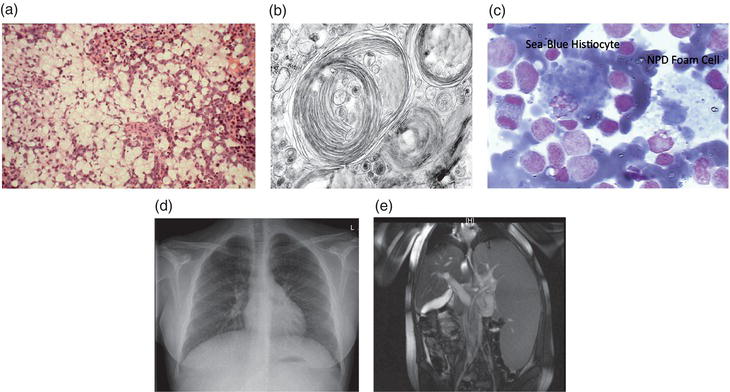
Figure 10.1 (a) Typical H & E staining of a liver section from an ASM-deficient NPD patient. Note the massive infiltration of lipid filled macrophages (foam cells). (b) Electron micrograph showing membrane-like inclusions in the brain of a type A NPD patient. (c) Bone marrow from a type B NPD patient showing an NPD foam cell and sea-blue histiocyte, both characteristic of ASM-deficient NPD. (d) Chest radiograph of a type B NPD patient showing the diffuse, reticulonodular interstitial changes. (e) High resolution CT showing hepatosplenomegaly in a type B NPD patient.
Related posts:





Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


