The canonical endocytic pathway progresses along an increasing lumen-acidic gradient from early endosomes retrogradely trafficked from the plasma membrane, to multivesicular bodies or late endosomes, and finally to perinuclear-localized lysosomes. Deviating from this pathway, early endosomes can be recruited back to the plasmalemma or to other organelles as sorting/recycling endosomes. These divergent streams allow for the recycling and reinsertion of cell surface receptors, delivery of signaling ligands throughout the cell, and internalization of membrane components to be reorganized. Such carefully orchestrated processing and its related signal transduction events may be interrupted in diseases of the lysosomal system in which endocytosed components including cholesterol and GSLs accumulate. While this accumulation is typically associated with late endosomes and lysosomes, recent evidence has emerged suggesting additional involvement of early endosomal compartments [3], raising the likelihood that the consequences of lysosomal disease extend well beyond the lysosome itself.
Autophagy
In addition to endocytic pathways, autophagic streams feed into the lysosome and are involved in targeting intracellular material including effete organelles, long-lived proteins and pathogens for degradation [4]. Autophagy, which is often activated following starvation stress, is divided into three distinct subtypes – microautophagy, chaperone-mediated autophagy (CMA) and macroautophagy – defined by the delivery method of substrate to lysosomes. Microautophagy involves the direct engulfment of cytosolic material by the lysosome either by direct invagination of the lysosomal membrane, or projected arm-like extensions that sequester material into intralysosomal vesicles. CMA is selective for soluble monomeric proteins containing the peptide signalling motif Lys-Phe-Glu-Arg-Gln (KFERQ). This motif allows for binding by the heat shock cognate 70 protein (HSC70). HSC70 and co-chaperones then promote protein unfolding and translocation across the lysosomal membrane via the lysosome-associated membrane protein type 2A (LAMP2A). Macroautophagy has been traditionally characterized as a vesicle-mediated bulk degradation mechanism activated in response to nutrient deprivation. Recently, however, it has been shown that macroautophagy is also constitutively active and selective. When macroautophagy is stimulated, double membrane vesicles known as autophagosomes form to engulf cytosolic material. This material, including dysfunctional organelles and oligomerized proteins, is selectively recognized by chaperone complexes and adapter molecules including heat-shock proteins (HSPs), ubiquitin, and p62/SQSTM1 (p62) which allow for the specific uptake of these substrates within autophagosomes. To date several lysosomal diseases have been found to exhibit alterations in autophagy, however the downstream consequences for disease progression remain unclear. Of particular interest is evaluating how impaired autophagy contributes to neurodegenerative processes.
Salvage
Degradation products resulting from lysosomal processing must be efficiently removed from the organelle for utilization elsewhere in the cell. This salvage process involves numerous lysosomal membrane proteins that act as transporters, including cystinosin, sialin, cobalamin transporter and NPC1 protein (see Chapter 17). For example, a defect in the salvage of cobalamin (also known as vitamin B12) leads to cobalamin F-type disease, a disorder characterized in children by megaloblastic anaemia, failure to thrive and neurological deficits [5]. The inability of cobalamin to leave the lysosome and enter the cytosol means that it is unavailable for conversion into enzyme cofactors methylcobalamin and adenosylcobalamin, which are critical for multiple metabolic processes. Another example of salvage compromise involves the reutilization of lysosomal degradation products directly in synthetic pathways in the Golgi and elsewhere in the cell. In Niemann–Pick type C disease (NPC), lack of the NPC1 protein and the resulting compromise in egress of unesterified cholesterol to other sites in the cell is known to cause compensatory increases in cholesterol synthesis [6] and a similar phenomenon involving glycosphingolipids (GSLs) may occur following sequestration of GM2 and GM3 gangliosides [3]. Following synthesis in the Golgi and transport to the plasmalemma, GM1 and other complex gangliosides are eventually endocytosed and degraded in lysosomes. Salvage of simple gangliosides (e.g., GM2 and GM3) from lysosomes and their transport back to the Golgi allows for efficient production of complex gangliosides without the need for complete de novo synthesis from ceramide. Even in other lysosomal disease caused by defects in degradation rather than egress – such as Sandhoff disease in which GM2 accumulates following absence of β-hexosaminidase – neurons may similarly be deprived of this precursor for complex ganglioside synthesis. Reutilization of simple molecular components derived from lysosomal degradation would be energetically favourable over full de novo synthesis. What storage may lead to in terms of cell energy consumption and altered regulatory processing is only now beginning to be explored in depth. Thus, while lysosomal diseases have been historically viewed as states of overabundance of non–degraded material, it is only logical to assume that they are also likely deficiency diseases in which critical components for multiple metabolic pathways are in reduced supply.
Ubiquitin–Proteasome System (UPS)
In addition to pathways directly feeding into and out of the lysosome, the lysosomal system includes mechanisms functionally allied in maintaining proteolytic quality control, namely the UPS. As the primary degradative pathway for most soluble short-lived proteins, the UPS plays a pivotal role in cellular regulatory processes, including the endoplasmic reticulum associated degradation system (ERAD). The ERAD system is responsible for turning over aberrantly misfolded proteins immediately following their translation thereby serving as a quality control check-point. Additionally, the UPS has been found to coordinate proteolysis with the autophagic/lysosomal system in certain instances. For example, UPS inhibition promotes the upregulation of macroautophagy in an apparent effort to redirect and sustain efficient protein degradation through the lysosome. Furthermore, the UPS and autophagic/lysosomal systems rely on several of the same key molecules – most notably ubiquitin and p62 – selectively targeting substrates for degradation. This complementation is limited, however, as the UPS only has the ability to degrade monomeric proteins that have been properly unfolded and fed into the proteasome catalytic core, while macroautophagy is capable of bulk degradation of organelles and oligomerized proteins. In lysosomal disease states in which the degradative capacity of the lysosome is compromised it is conceivable that the UPS may be employed to compensate for some of the proteolytic load, although the molecular machinery and sequence of events involved in this cross-talk is largely unknown. Finally, it is unclear whether there is a threshold at which the UPS may too become overwhelmed, perhaps contributing to ER stress and thereby causing complete proteolytic failure in lysosomal disease states.
Lysosomal diseases
After more than a half century of clinical recognition and classification, lysosomal storage diseases were conceptualized as disorders resulting from deficiencies in single lysosomal hydrolases followed by the subsequent accumulation of a specific substrate for that enzyme [7]. Today, lysosomal diseases are known to include nearly 60 monogenic disorders with a combined frequency of approximately 1: 7000 live births. They are known to be caused by deficiencies not only in lysosomal hydrolases, but also in other lysosomal and non-lysosomal proteins including enzymes, soluble non-enzymatic proteins, and membrane-associated proteins critical for proper function of the greater lysosomal system [8]. Categorizing lysosomal disorders is not completely straightforward as several diseases exhibit significant overlap of pathological features, storage material, and so forth. However, grouping lysosomal diseases based on the traditional biochemical nature of the primary storage material is often preferred; these include the lipidoses, mucopolysaccharidoses (MPSs), glycogenoses, neuronal ceroid lipofuscinoses (NCLs), mucolipidoses (MLs), glycoproteinoses and others (see Classification in Chapter 5).
In general, most lysosomal diseases afflict children. Age of onset and clinical course can vary significantly, but nearly all lysosomal diseases have a delayed non-congenital onset, and progressive course ultimately leading to premature death. Even as our grasp of the genetic, molecular and biochemical bases of these disorders has advanced in recent years there are still many gaps in our understanding as to pathogenic mechanisms and the reversibility of disease-induced cell damage. Some of the most important questions in this regard involve the brain.
CNS Involvement
(See also Chapter 21) Adding to lysosomal disease complexity is the broad systemic involvement of multiple tissues and organs. Clinical presentation often involves bone, muscle, liver, kidney and spleen, while nearly two-thirds of these disorders also exhibit extensive neurological impairment (see Chapter 2). Intellectual disability, dementia, seizures, motor system deficits, visual impairment and hearing loss are common manifestations associated with several lysosomal diseases. The progressive clinical and pathological course presented in these disorders highlights the indispensible role of the lysosome, especially in post-mitotic neurons which are predominately affected. Interestingly, this vulnerability becomes manifest in a highly specified manner with different neuronal subtypes and brain regions exhibiting distinct pathophysiological changes.
One of the more perplexing phenomena associated with lysosomal disease is neurodegeneration. While many of these disorders exhibit some degree of patterned neuronal loss, the question remains why some neurons appear more vulnerable to this fate than others and what effect neurodegeneration has on disease course. A common theme implicated in motor system impairment is Purkinje cell (PC) death. PC death in mice with NPC begins early and progresses in an anterior to posterior lobe pattern within the cerebellum. This cell loss is severe and nearly total, with the conspicuous exception of lobule X (flocculonodular lobe in humans) where PCs are well preserved. Remarkably, almost identical patterns of PC loss occur across a wide spectrum of lysosomal diseases, including mucolipidosis type IV and the gangliosidoses. It should also be noted that in the case of NPC disease that both PCs and cerebrocortical neurons exhibit extensive pathological storage of cholesterol and ganglioside, however unlike PC degeneration, cortical neuron death is often not as conspicuous in the early stages of the disease.
CNS inflammation is an additional feature of lysosomal diseases that in many cases has been shown to be spatially and temporally correlated with neuronal dysfunction and neurodegeneration. Of particular interest is how this initial protective response to disease may become deleterious with chronic induction, ultimately leading to secondary damage and exacerbation of pathogenic processes. In the normal brain, microglia play an essential house-keeping role intimately coupled to neuronal function. As the resident macrophage of the CNS, microglia are also critical for synapse maintenance, axon and spine pruning, clearing extracellular debris and apoptotic cells, glutamate and trophic factor regulation, and probing their surroundings for homeostatic deviations in the extracellular environment. Conversely, during disease/injury activated microglia proliferate, and, along with infiltrating macrophages, can generate cytotoxic components including reactive oxygen species, nitric oxide and pro-inflammatory cytokines. Microglia and macrophages exhibit altered morphological states in many lysosomal diseases, while activated astrocytes are also a prominent feature contributing to the pathological landscape in the CNS. Some NCLs, gangliosidoses, MPSs, MLs and neuronopathic Gaucher disease exhibit neuroinflammatory features. A study in Sandhoff disease mice showed that microglial activation precedes neuronal cell death, and that bone marrow transplantation ameliorates the expansion of microglia and neurodegeneration [9]. Interestingly, however, this improvement did not coincide with any significant decrease in GM2 ganglioside storage, suggesting that microglial activation is a significant component of neuronal death independent of storage. As such, the use of anti-inflammatory drugs to treat lysosomal diseases may be a relevant therapeutic strategy for providing benefit in certain instances. Future efforts to decipher the protective roles of microglial activation and inflammation from pathogenic stimulating events, and to determine the temporal window for using anti-inflammatory drugs in the treatment of lysosomal disease states, will be critical.
Intracellular storage
Ever since the concept of lysosomal disease was developed by H.G. Hers, intracellular storage material has been a defining characteristic of these disorders. As the complexity of this pathological feature has evolved well beyond the original single enzyme/single substrate theory, research has focused on understanding how storage contributes to pathogenic cascades. The relationship between primary and secondary storage warrants clarification. Primary storage may be defined as the lysosomal buildup of biochemical components that accumulate as a direct result of a failure of degradation within, or egress of, degradation products from the lysosomal system. Secondary storage is material accumulating from subsequent downstream compromise in lysosomal function. Given a number of lysosomal diseases caused by proteins of unknown or poorly understood function, however, the classification of specific storage material as primary or secondary continues to evolve. Neuronal storage is often found in perinuclear regions of the perikarya, although it may extend into dendrites and the axon hillock. Large swellings in the latter are known as meganeurites (described below).
At its simplest, the buildup of primary storage material probably further compromises the degradative capacity of lysosomes and thereby exacerbates the accumulation of secondary storage components in a deleterious cycle. More complex is the question of how storage may affect the lysosomal system as a whole through such expanding downstream events. These include the regulation of appropriate lysosomal pH, proper coordination of fusion events between autophagosomes, endosomes and lysosomes, unabated signal transduction along the endocytic pathway, and efficient and appropriate lysosomal salvage for regulating biosynthetic processes. Like selective neurodegeneration, differences in storage can also occur in distinct neuronal populations with regional specificity. For example, in mucolipidosis (ML) IV mice, different gangliosides accumulate throughout the CNS; in the hippocampus, however, gangliosides appear to be confined to specific regions – storage of GM3 occurs in the CA3 region, while GD3 is found in the CA1–CA2 regions only [10]. What this region-dependent storage represents remains unknown.
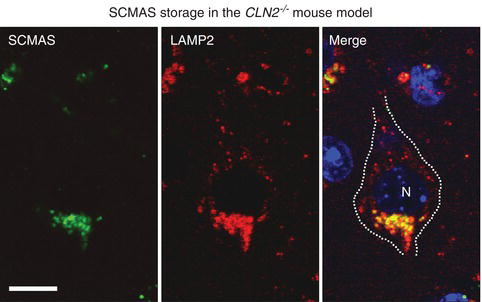
Determining whether lysosomal storage per se is a fundamental cause of neuronal dysfunction remains an important question. Studies in NCL disease have shown no direct correlation between the accumulation of saposins A and D and subunit c of mitochondrial ATP synthase (SCMAS) and neuron loss (Figure 1.2). In fact, storage pathology in NCL and other lysosomal diseases is typically prevalent long before the onset of any behavioural phenotype in animal models [8]. Similarly, neurons in many lysosomal diseases present significant storage pathology early, and yet survive for extended periods of time (decades) suggesting that accumulation is not immediately cytotoxic. Yet this does not mean that neurons remain functionally normal. Indeed, as described below, given the presence of metabolic compromise and of axonal, dendritic and synaptic abnormalities, it is highly likely that affected neurons are not optimally functioning even from early time points in the storage process. Eventually the presence of such variously malfunctioning neurons in given neural networks would be anticipated to reach a tipping-point at a systems level, with clinical disease emanating as a result, even in the absence of frank neurodegeneration. Such clinical deficits would be solidified, and possibly worsened, with the eventual death of neurons that participate in these neural circuits. It is essential to understand these issues in the face of emerging therapies that may rescue the storage phenotype long after it has been established, but before neuron death. Interestingly, substrate reduction therapy either aimed at inhibiting the biosynthesis of GSLs in gangliosidoses [11], or at enhancing the egress of cholesterol and GSLs in NPC disease, has proven effective in delaying clinical onset and increasing life-span [12]. These studies clearly indicate that reducing storage is beneficial in lysosomal disorders but reveal little about the precise link between storage and brain dysfunction.
Figure 1.2 Storage accumulation of subunit c of mitochondrial ATP synthase (SCMAS) in a cortical pyramidal neuron of the Cln2 -/- mouse model of classiclate-infantile NCL disease. Image by confocal immunofluorescence microscopy of a single plane from a z-series showing SCMAS colocalization with the late endosomal/lysosomal marker LAMP2. Merged image stained with the nuclear marker DAPI (N;nucleus). Scale bar represents 10 μm.
Related posts:





Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


