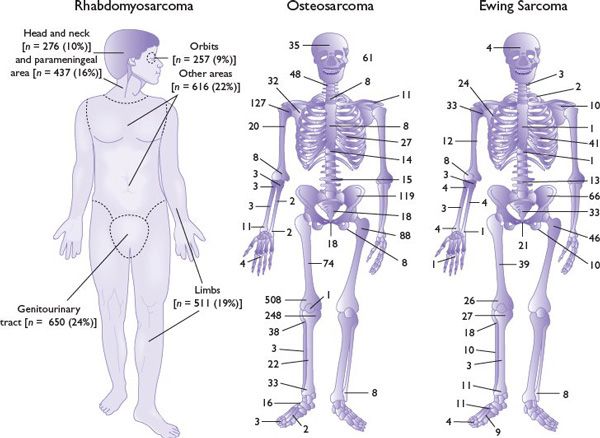
FIGURE 21.1 Primary sites of rhabdomyosarcoma, osteosarcoma, and Ewing sarcoma, showing numbers of patients with primary tumors at specific sites.
Pathophysiology
RMS is of mesenchymal origin and is characterized by myogenic differentiation. There are two main histologic subtypes, embryonal (80%) and alveolar (15% to 20%), with characteristic genetic differences (Table 21.2 and Fig. 21.2. Botryoid RMS and spindle cell sarcoma are both morphologic variants of embryonal RMS. Numerous environmental and genetic factors have been associated with an increased risk of RMS (Table 21.3).
Diagnosis
Radiologic
Comprehensive radiologic evaluation includes the following:
■Tumor localization
•Computerized tomography (CT) and magnetic resonance imaging (MRI)
•Positron emission tomography (PET)
■Assessment of metastatic spread
•CT of chest/lungs
•Technetium bone scan for bone or bone marrow involvement
•PET

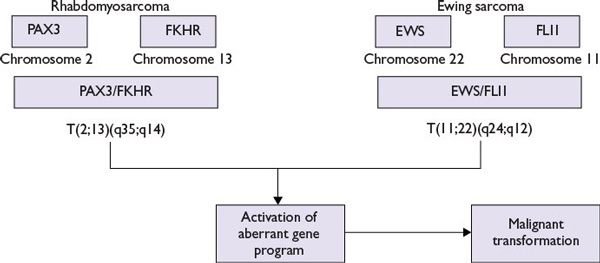
FIGURE 21.2 Molecular pathogenetic mechanisms in rhabdomyosarcoma and Ewing sarcoma.
Pathologic
Open biopsy is the preferred approach for tissue diagnosis and should be undertaken at an oncology center, where diagnostic material can be optimally used and the initial surgical approach can be determined by a multidisciplinary team responsible for the patient’s subsequent treatment. Needle biopsy may restrict access to fresh and frozen tissue for cytogenetic and molecular genetic investigations.
1.Tumor characterization
•Histopathology
•Immunohistochemistry (desmin and myoD1)
•Genetics
•RT-PCR for presence of PAX/FKHR translocation
•Cytogenetics
2.Metastatic spread
•Cerebrospinal fluid PCR for PAX/FKHR translocation
•Bone marrow aspirate cytology
•Bone marrow biopsy for histochemistry and PCR
Treatment
The diversity of primary sites, distinctive surgical approaches and radiotherapies for each primary site, subsequent site-specific rehabilitation, and potential treatment-related sequelae (Table 21.4) underscore the importance of treating children and young adults with RMS in the context of a clinical trial at a major medical center that has appropriate experience in all therapeutic modalities.

Surgery
Local tumor control is the cornerstone of therapy, especially for patients with nonmetastatic disease. Primary tumor resection should be undertaken only if there is no evidence of lymph node or metastatic disease and if the tumor can be excised with good margins without functional impairment or mutilation. Surgery has minimal, if any, role in the primary management of orbital tumors and a limited role in local control of head and neck tumors. To achieve local control of pelvic tumors in very young children, the risks of radical surgery may be more acceptable than those of pelvic irradiation.
Radiotherapy
Radiotherapy after initial surgical resection or chemotherapy is recommended in the following instances:
■Completely resected tumor (clinical group I) with unfavorable histology (alveolar RMS)
■Microscopic residual disease (clinical group II: up to 4,100 cGy)
■Gross residual disease (clinical group III: up to 5,040 cGy)
Treatment Volume
■Volume is determined by extent of disease at diagnosis
■Radiation field should extend 2 cm beyond the tumor margin
■Whole-brain irradiation of 2,340 to 3,060 cGy for parameningeal disease with intracranial extension
Chemotherapy
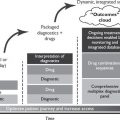
Neoadjuvant combination, multiagent chemotherapy for extensive, primarily unresectable tumors is known to reduce the extent of subsequent surgery or radiotherapy. (Fig. 21.3 outlines this multidisciplinary approach for RMS.)
OSTEOSARCOMA
Osteosarcoma is a primary bone malignancy with peak incidence in the pubescent growth spurt (15 to 19 years) in the metaphyses of the most rapidly growing bones. Risk factors are listed in Table 21.5 and clinical presentation in Table 21.6.
Clinical Presentation
■Bone pain
■Swelling
■Mass in metaphyseal area of bone, most commonly femur or tibia
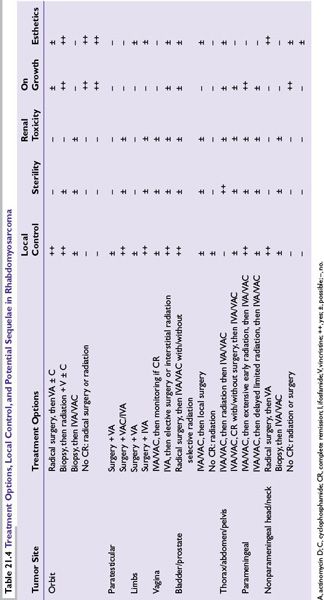
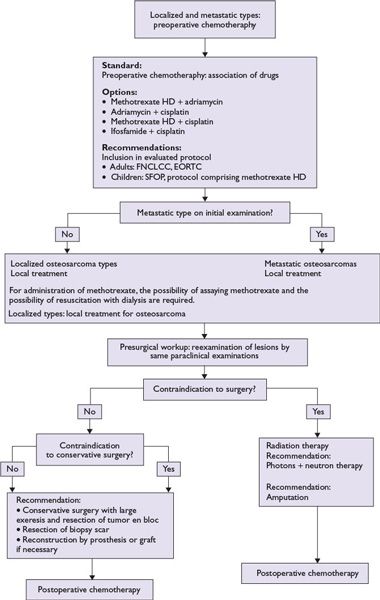
FIGURE 21.3 Treatment options for osteosarcoma.


Diagnosis and Staging
Radiologic
■Tumor assessment: plain radiographs
•Destruction of bone with consequent loss of normal trabeculae and appearance of radiolucent areas
•New bone formation
•Lytic or sclerotic appearance
•“Sunburst sign”: periosteal elevation from tumor penetrating cortical bone
■Extent of disease
•MRI (T1-weighted) to assess primary tumor boundaries in entire long bone, including skip lesions
•Technetium bone scan
•PET
■Metastatic spread (15% to 20%)
•Technetium bone scan
•CT of chest/lungs
•PET
Pathologic/Genetic
■
Related posts:


Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


