Prognosis
■Prognosis varies by thyroid cancer subtype, but the overall 5-year relative survival is nearly 98%. This is because more than 80% of cases are papillary thyroid cancer (PTC), the subtype with the best survival.
Differentiated Thyroid Cancer: Papillary, Follicular, Hurthle Cell
■More than 90% of all thyroid cancers are a subtype of differentiated thyroid cancer (DTC).
■PTC is the most common subtype (80% to 85%).
■PTC is generally unilateral, but may be multifocal within a lobe. Variants include tall cell, columnar, and diffuse sclerosing.
■PTC metastasizes primarily via lymphatic invasion; vascular invasion is uncommon.
■Genetic alterations involved in the MAPK signaling pathway are found in at least 75% of PTC cases. BRAFV600E mutation is found in approximately 45% of PTCs, while RET rearrangements are found in approximately 25% Activating point mutations in the RAS oncogenes occur in approximately 10% of cases.
■Follicular thyroid cancer (FTC) is the second most common type of thyroid carcinoma, comprising 10% to 15% of thyroid cancers.
■FTC typically disseminates hematogenously, with metastases to bone and lung being most common in advanced disease.
■RAS point mutations and the PAX8/PPARγ translocation are the most common genetic alterations in FTC.
■Hurthle cell cancer (HCC) is also referred to as oxyphilic or oncocytic thyroid cancer, and represents approximately 5% of all DTCs. It is often considered a variant of FTC with less sensitivity to radioiodine and a more aggressive clinical course.
Clinical Presentation
■Most patients present with an asymptomatic thyroid nodule. Clinical symptoms may include the following:
•Hoarseness caused by invasion of the recurrent laryngeal nerve or by direct compression of the larynx
•Cervical lymphadenopathy
•Dysphagia
•Horner syndrome (miosis, partial ptosis, hemifacial anhidrosis)
Diagnosis
■Evaluation of any suspected thyroid nodule >1 cm should include a serum TSH and thyroid ultrasound. If a nodule is seen on ultrasound:
•If TSH is normal or high, a fine-needle aspirate (FNA) should be done.
•If the TSH is low, the nodule should be evaluated by radionuclide scan to see if it is hyperfunctioning. Hyperfunctioning nodules are benign and patients with them should be treated for hyperthyroidism.
■Up to 30% of FNAs are indeterminate; therefore, a definitive diagnosis is often not made until the nodule is resected. A new gene expression classification assay was able to predict benign pathology when FNA cytology was indeterminate, and may allow a more conservative approach for those who would otherwise undergo a diagnostic surgical procedure.
■Carcinoma is suggested by the following clinical findings: a history of head and neck radiation, family history of thyroid cancer, exposure to ionizing radiation, rapid growth of the nodule, hoarseness, vocal cord paralysis, and lymphadenopathy. There may also be specific features on ultrasound that are suggestive of possible malignancy.
■Staging for DTC incorporates age. For patients ≤45 years old, the most advanced they can be is stage II given their excellent prognosis.
Treatment
Surgery
■Total thyroidectomy is recommended for a DTC lesion >1 cm, a lesion that extends beyond thyroid, or for patients with history of prior exposure to ionizing radiation to head/neck.
■Unilateral lobectomy with en bloc resection of tumor may be considered for a DTC lesion <1 cm or for follicular lesion with no evidence of multicentric disease.
■Modified radical neck dissection should be done for regional lymph node metastases.
■Thyroidectomy should be performed in patients with distant metastases to permit treatment with radioiodine, which can still be curative.
Adjuvant Therapy
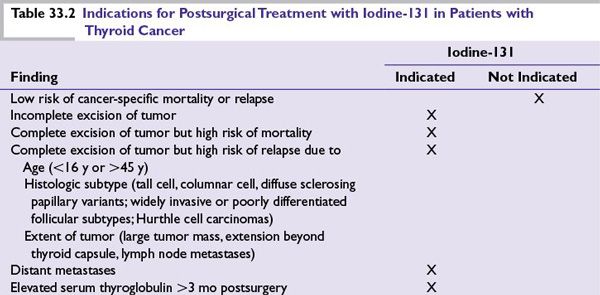
■Treatment with radioiodine (I-131, RAI) is used to ablate normal residual thyroid tissue, treat micrometastases, and decrease cancer-related death, tumor recurrence, and development of distant metastases. Table 33.2 outlines indications for iodine-131 treatment after surgery.
■Adjuvant external beam radiotherapy is sometimes recommended for those patients with gross or microscopic residual disease or those with high-risk histology and visible extrathyroidal extension. Locally recurrent disease not amenable to surgery or radioiodine therapy can also be treated with external beam radiotherapy.
Targeted Therapy/Chemotherapy
■Patients with advanced disease that is refractory to I-131 and unresectable should participate in a clinical trial when possible. These patients are often treated with a small molecule VEGFR inhibitor (off label) based on phase II studies that have shown responses and clinical benefit. Traditional systemic chemotherapy has largely been ineffective. Isolated metastases can be treated with external beam radiotherapy.
Medullary Thyroid Cancer
■Medullary thyroid cancer (MTC) is a calcitonin-secreting tumor of the parafollicular (C) cells occurring either sporadically (75% of cases) or as a part of hereditary syndrome (25% of cases). The hereditary syndromes are the multiple endocrine neoplasia type 2 syndromes (MEN 2A or 2B) and familial medullary thyroid cancer (FMTC) (see Table 33.3).
■MEN 2 and FMTC are autosomal dominant syndromes caused by germline RET oncogene mutations. Somatic RET mutations also occur in approximately 50% of sporadic MTC.
■MTC represents 3% to 5% of all thyroid cancer cases.
■Sporadic tumors tend to be solitary, whereas familial tumors tend to be bilateral and multifocal.

Clinical Presentation
■Patients typically present with an asymptomatic thyroid mass. Some may also have local symptoms such as dysphagia, dyspnea, or hoarseness.
■Approximately 10% will present with systemic symptoms usually consisting of bone pain, flushing, and/or diarrhea.
■Approximately 50% of patients present with regional lymphadenopathy.
■Distant metastases typically occur in late-stage disease and usually involve lung, liver, bones, and adrenal glands.
Diagnosis
■Guidelines for evaluation of thyroid nodules should be followed as described for DTC.
■If the FNA is suggestive of MTC, further evaluation should consist of calcitonin and CEA measurement and genetic testing for germline RET mutations.
Treatment
■Total thyroidectomy with central lymph node dissection is the appropriate surgery.
■Surgery and/or external beam radiotherapy can be used for residual or recurrent disease treatment; however, the survival benefit for either modality is unclear.
■Patients with advanced, progressing, or symptomatic residual or recurrent disease not appropriate for surgery or radiation therapy should be considered for systemic therapy.
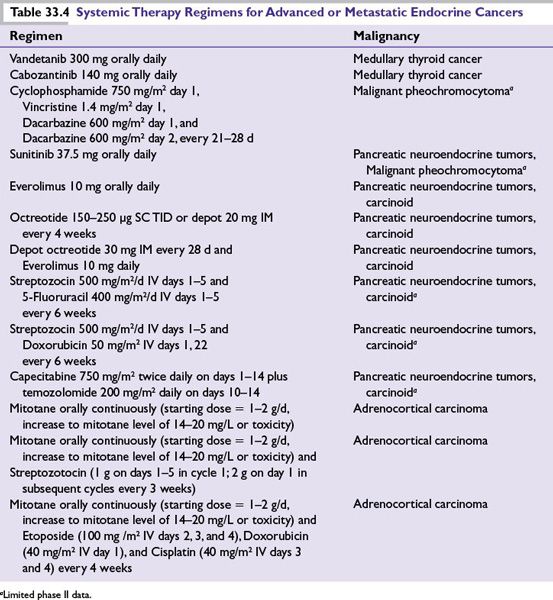
■Both vandetanib and cabozantinib have recently been approved by the US FDA for the treatment of advanced MTC based on improvement in progression-free survival in phase III trials. No improvement in overall survival has been demonstrated; therefore, patients with indolent disease should consider observation until their disease becomes necessary to treat. See Table 33.4 for vandetanib and cabozantinib dosing.
■Traditional systemic chemotherapy has largely been ineffective.
Anaplastic Thyroid Cancer
■Anaplastic thyroid cancer (ATC) is a rare, high-grade, aggressive malignancy that accounts for 2% to 5% of all thyroid carcinomas. Up to 50% of patients have antecedent or concurrent history of DTC. Disease-specific mortality is nearly 100%.
■Patients typically present with a rapidly enlarging neck mass.
■Approximately 90% will have locoregional or distant metastases at the time of diagnosis.
■Treatment is primarily palliative and often aimed at preventing asphyxiation, the most common cause of death in these patients. It can consist of surgery, radiation, chemotherapy, or a combination of these modalities.
Other Thyroid Cancers
■Primary thyroid lymphoma
■Metastasis to the thyroid
PARATHYROID CARCINOMA
Clinically, it is important to distinguish this disease from other benign disorders that cause hyperparathyroidism. Parathyroid carcinoma accounts for less than 1% of cases of hyperparathyroidism.
Epidemiology and Natural History
■Parathyroid carcinoma occurs in <1 per million individuals per year, predominantly diagnosed in the fifth or sixth decade of life.
■Germline or somatic mutations of the HRPT2 tumor suppressor gene are detected in the majority of cases.
■Ten-year survival rate is approximately 70%; however, 40% to 60% will recur after initial surgery.
■Morbidity and mortality are usually related to hypercalcemia rather than complications of metastases.
Clinical Presentation
Patients typically present with the following:
■Symptoms of hypercalcemia, with calcium levels usually >14 mg/dL
■Elevated parathyroid hormone levels
■Palpable neck mass in up to 70%
■Metastases to cervical lymph nodes, lungs bone, or liver in approximately 10%
Diagnosis
■Parathyroid carcinoma is difficult to diagnose preoperatively; differential includes parathyroid adenoma and hyperplasia.
■Most parathyroid carcinomas are diagnosed at surgery; however, some are not diagnosed until local recurrence or metastases. This is because there are no definitive histopathologic features to differentiate carcinoma from adenoma.
■FNA is inappropriate for diagnosis.
Treatment
Surgery
■Treatment consists of parathyroidectomy with en bloc resection of tumor and involved structures. This may include the ipsilateral lobe of thyroid. Radical lymph node dissection is not recommended.
■Recurrent tumor and oligometastases should also be resected.
Radiation
■Parathyroid tumors are generally not radiosensitive.
■Small retrospective studies suggest there may be improved local control with postoperative radiotherapy for high-risk patients.
■Radiation may have palliative benefit.
Medical Therapy
■Chemotherapy efficacy is limited to case reports, and there is no standard regimen.
■Management of hypercalcemia is essential while treating parathyroid carcinoma.
ADRENOCORTICAL CARCINOMA
Epidemiology
■ACC is a rare malignancy arising from the adrenal cortex, with 1 to 2 cases per million population per year.
■It has a bimodal age distribution, with a first peak in children younger than 5 years and a second peak in adults in their fourth to fifth decade.
■
Related posts:



Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


