The most common signs in order of decreasing frequency are
■Hemiparesis
■Cranial nerve palsies
■Papilledema
■Cognitive dysfunction
■Sensory deficits
■Hemiparesthesia
■Hemianopia
■Dysphasia
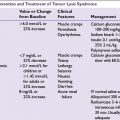
Acute Complications
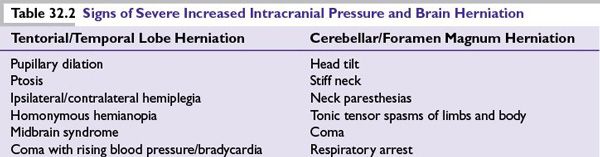
Because the skull’s rigidity does not allow for intracranial expansion, brain lesions can result in structural displacement and life-threatening consequences. Following the path of least resistance, increased intracranial pressure (ICP) may cause tentorial or foramen magnum herniation, causing significant neurologic signs (Table 32.2).
Types of Primary Brain Tumors
Gliomas
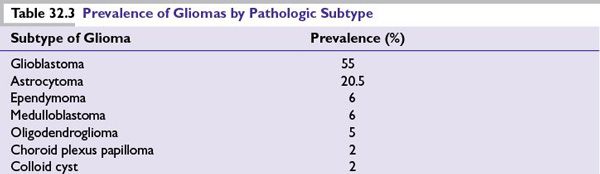
Gliomas account for approximately 65% of all intracranial tumors (range 46% to 85% in various databases), with age-related incidence by histologic subtype. Table 32.3 shows the prevalence of the pathologic subtypes of gliomas in relation to other more common primary brain tumors.
There are four major types of gliomas, based on their presumed glial cell of origin:
■Astrocytoma (glioblastoma multiforme [GBM], anaplastic astrocytoma, diffuse astrocytoma, pleomorphic xanthoastrocytoma, pilomyxoid astrocytoma, pilocytic astrocytoma, giant cell astrocytoma)
■Oligodendroglioma (anaplastic oligodendroglioma, oligodendroglioma)
■Mixed glioma (anaplastic oligoastrocytoma, oligoastrocytoma)
■Ependymoma (anaplastic ependymoma, ependymoma, subependymoma, myxopapillary ependymoma)

Grading The World Health Organization’s pathologic grading system, generally accepted by neuropathologists since 1993 (most recently updated in 2007), determines the grade (level of aggressiveness) of each histologic subtype of tumor (Table 32.4) based on the following features:
■Cellular atypia
■Mitotic activity
■Degree of cellularity
■Endothelial proliferation
■Degree of necrosis and/or microvascular proliferation
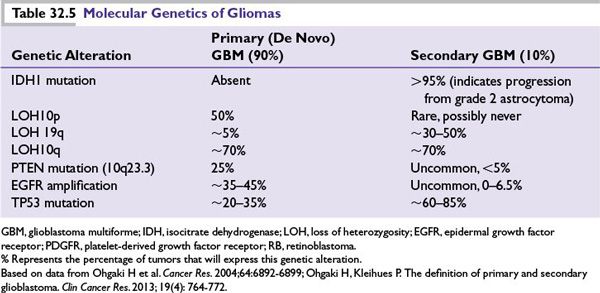
Molecular Genetics Patterns of genetic abnormalities have been identified in various glioma subtypes (Table 32.5). Secondary or progressive gliomas often demonstrate mutations of p53, but they seldom show amplification of epidermal growth factor receptor (EGFR). By contrast, primary (de novo) GBMs usually lack p53 mutations and contain an amplified EGFR. IDH (isocitrate dehydrogenase) mutations appear to be pathognemonic for secondary GBM diagnosis by current definitions of the two entities, which indicate common precursor events for all secondary GBMs.
Glioblastoma Multiforme (WHO Grade IV Astrocytoma)
■The most common adult primary brain tumor, accounting for about half of all gliomas and 10% to 15% of all intracranial tumors.
■Peak incidence is at 45 to 65 years; overall incidence is 2 to 3/100,000; male-to-female ratio is 3:2; median survival is around 3 months if untreated, approximately 12 to 15 months with standard therapy (discussed below).
■More likely to cross the corpus callosum than other types of brain tumors.
■Development may occur de novo (primary) or after progression from a lower-grade precursor lesion (secondary).
Imaging Characteristics
■Heterogeneous hypointense or isointense mass on CT or T1-weighted MRI.
■Heterogeneously contrast-enhancing mass, often the presence of necrosis visible.
■Hypervascular appearance.
■Rare calcifications (more common in oligodendroglial tumors).
■MR spectroscopy is increasingly used to distinguish tumor from other processes visualized on MRI.
■Gliosarcoma, a variant, has a mesenchymal component and a greater tendency for dural invasion.
■GBMs are characteristically infiltrative within brain parenchyma but rarely show extracerebral metastasis.
■High-grade tumors (by definition, GBM) are commonly hypermetabolic on FDG PET.
■Single-photon emission-computed tomography (SPECT) and MR cerebral perfusion imaging may distinguish radiation necrosis (hypovascular) from tumor recurrence (hypervascular).
Differential Diagnosis
■Brain metastasis
■Cerebral abscess
■Demyelinating/inflammatory process (i.e., multiple sclerosis)
■Radiation necrosis
Treatment
First Line
■Maximal surgical debulking is of primary importance after diagnosis is established (not ideal if primary CNS lymphoma is suspected). The degree of resection is a strong prognostic factor.
■The landmark study, which established the standard of care for upfront therapy, was a randomized phase 3 trial by the EORTC-NCIC (European Organisation for Research and Treatment of Cancer and National Cancer Institute of Canada Clinical Trials Group), reported by Stupp et al. in the New England Journal of Medicine in 2005.
■This study randomized 573 patients between August 2000 and March 2002 to radiotherapy (focal, 60 Gy in 2 Gy fractions given 5 days per week over 6 weeks) alone or in combination with temozolomide (75 mg/m2, PO 7 days per week during concurrent chemotherapy, then 150 to 200 mg/m2 PO days 1 to 5 of 28-day cycles for up to six cycles). Patients were stratified according to performance status, previous surgical intervention (yes/no and % resected), and treatment center.
■Concurrent chemotherapy and radiotherapy resulted in an improved median overall survival (OS) (14.6 months) compared with radiotherapy alone (12.1 months). The 2-year survival rate also favored the combination arm (26.5% vs. 10.4%) with minimal additional toxicity.
■An analysis of the tumor tissue identified methylation of the MGMT promoter as an excellent indicator of response and prognosis with temozolomide. In the combined group, patients with MGMT promoter methylation had a median OS of 23.4 months compared with 12.6 months for the patients without promoter methylation.
■Gliadel wafers (3.8% carmustine) are also FDA-approved for newly diagnosed GBM, which are placed locally into the tumor bed during surgery, followed by radiotherapy for newly diagnosed GBM (increased median OS by 2.1 months vs. radiotherapy alone). This approach carries an increased risk of CNS leakage (can cause increased infections and increased need for use of steroids to treat swelling).
Second-Line or Other Agents
■Bevacizumab (Avastin, monocolonal antibody against circulating VEGF) given 10 mg/kg IV over 30 to 90 minutes every 2 weeks—capable of inducing significant tumor responses in phase 2 studies for recurrent GBM.
■Other standard agents used to treat malignant gliomas are generally alkylating agents and include irinotecan, carboplatin, procarbazine, etoposide, carmustine, and lomustine.
■Other promising strategies using inhibitors of the EGFR, PDGFR, ras, and mTOR (mammalian target of rapamycin) signal transduction pathways are being studied. Additional treatment strategies currently under investigation include therapeutic gene transfer and immunotherapeutic approaches.
Prognosis
■See Table 32.6—based on several Radiation Therapy Oncology Group trials.
Astrocytoma and Anaplastic Astrocytoma
Low-Grade Diffuse Astrocytoma Low-grade diffuse astrocytomas are categorized as grade 2 and account for approximately 5% of primary brain tumors. They occur mostly in the cerebral hemispheres, but may also occur in the brain stem. Median age at diagnosis is 35 to 45.
Imaging characteristics on MRI are most commonly as follows:
■A nonenhancing lesion on T1-weighted images after contrast
■A well-delineated hyperintense lesion with little edema/mass effect on T2-weighted images
■Difficult to distinguish from nonmalignant infarct/cerebritis/demyelination
■Rare calcifications
Treatment
■Maximal surgical debulking is recommended in all cases that can be performed safely except in the case of small, asymptomatic lesions, which may be observed radiographically until resection is required.
■“High-risk” patients (two or more of age > 40, KPS < 70, tumor dimension > 6 cm, tumor crossing midline, preoperative neurologic deficit) are usually recommended to receive postoperative radiotherapy (54 Gy, based on the EORTC study indicating equivalent efficacy with improved safety compared with 60 Gy). Low-risk patients (those not meeting criteria for high risk) may be considered for observation alone.
■RTOG 98-02 is a prospective randomized clinical trial in low-grade gliomas evaluating the use of chemotherapy. It randomized patients with astrocytoma, oligodendroglioma, or oligoastrocytomas, stratified by age, histology, performance status, and extent of resection, to RT alone or in combination with PCV (procarbazine, CCNU, and vincristine) chemotherapy. The addition of chemotherapy improved median PFS, but not OS, but in the subset of patients who survived >2 years, median OS was significantly improved, as was PFS. This finding indicates a delayed benefit for chemotherapy and indicates that it might be most beneficial for the patients with best prognosis.
■Pilocytic astrocytomas, a subset of low-grade astrocytomas, are the most common pediatric astrocytic tumors and virtually the only type curable with complete surgical resection.

Prognosis The number of poor prognostic factors influences prognosis for a given patient: age, location (resectablity influenced by surrounding structures), size, and characteristics (size, enhancement on MRI) of the tumor as well as its molecular profile (IDH mutation, 1p and/or 19q mutations, which confer improved prognosis). These were combined in a recursive partitioning analysis (RPA), which separated patient characteristics into prognostic signs and then used those signs to separate patients into RPA classes (I to VI initially) with lower numbers having improved prognosis. These classes have been slightly modified over the years, but are still an excellent prognostic tool for any patient with a newly diagnosed glioma.
As surgical techniques improve, overall prognosis improves because the patients with the worst outcomes do much better with excellent resections. Prognosis of a given patient is strongly influenced by the surgical expertise, and difficult cases should be referred to centers of excellence when possible for best outcomes.
Median survival for all patients:
■Five years: 65% to 85%
■Ten years: 25% to 50%
High-Grade Diffuse (Anaplastic) Astrocytoma High-grade diffuse astrocytomas are categorized as grade 3 and account for approximately 5% of primary brain tumors. Age at diagnosis is most commonly 35 to 55. They are distinguished from low-grade diffuse astrocytomas by increased mitoses. They have a high propensity for transforming into GBM. Survival is 2 to 5 years.
Treatment
■Treat essentially identically to GBM.
■Maximal surgical debulking with or without carmustine (BCNU) wafer implantation when possible followed by adjuvant therapy (concurrent metronomic temozolomide plus radiotherapy followed by temozolomide as discussed in GBM treatment).
Oligodendroglioma and Oligoastrocytoma (Mixed Glioma)
These diffuse cerebral tumors often appear with prominent areas of calcification on CT scan. They account for 5% to 10% of all gliomas and may have a better prognosis than astrocytomas. Like astrocytomas, low-grade oligodendrogliomas may progress to a higher grade.
Treatment
■For all grades, when safe to do so, a maximal surgical resection is the initial step in management, allowing confirmation of tumor grade and the use of more directed therapy as needed.
■When only subtotal resection is available or surgery cannot be undertaken at all, patients are treated depending on risk stratification as they would be in the adjuvant setting after maximal resection.
■High-risk patients (2 or more of the following risk factors: >40 years, KPS <70, tumor >6 cm, tumor crossing midline, more than minor neurologic symptoms preoperatively, one or no deletions of 1p and 19q, or IDH not mutated) are usually treated with adjuvant radiotherapy and, in many centers, chemotherapy (a randomized trial is evaluating the role of RT alone or in combination with PCV chemotherapy; temozolomide).
■Low-risk patients (with 1 or less high-risk feature) may be observed at patient preference, but are commonly treated with RT adjuvantly. Chemotherapy may also be considered depending on the likelihood of chemosensitivity of the tumor (may be dependent on the presence of codeletion of 1p and 19q).
■Radiation therapy has been the treatment of choice for low-grade progressive and anaplastic oligodendrogliomas. However, chemotherapy with PCV or temozolomide is occasionally used as neoadjuvant therapy to delay radiation therapy with its potential long-term neurotoxicity.
Ependymoma
Ependymomas comprise a spectrum of tumors ranging from aggressive childhood intraventricular tumors to low-grade adult spinal cord lesions. Typical locations are on the ventricular surface and the filum terminale.
Epidemiology
■Ependymomas account for 2% to 10% of all CNS neoplasms.
■Seventy-five percent of ependymomas are low grade.
■Fifty percent occur before the age of 5 years.
■Intracranial tumors are 60% infratentorial, 40% supratentorial, with 50% intraventricular.
■Overall incidence of spinal seeding is approximately 7% to 15.7% for high-grade infratentorial lesions and increases with uncontrolled primary lesions.
■Highly variable biologic behavior despite pathologic appearance.
Imaging
CT and MRI are highly suggestive of the presence of ependymoma (e.g., calcified mass on the fourth ventricle) but are not diagnostic.
Treatment
■Complete resection can be curative. Subtotal resection benefit is not clearly defined, but is often a standard practice prior to radiotherapy as mainstay of treatment.
■When complete surgical resection is achieved, CSF analysis and MRI of the spine are commonly performed to rule out evidence of spinal seeding.
■Observation and limited-field adjuvant radiation therapy are reasonable options for low-grade ependymomas with no evidence of metastases.
■Subtotal resection with no evidence of metastases should have limited field radiotherapy in the setting of tumor progression.
■
Related posts:


Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


