Hereditary Tumor Syndrome
Manifestation of Tumor
Gene (chromosomal locus)
Syndrome
Prevalence of ACT
Li-Fraumeni
TP53 (17p13), hCHK2 (22q12.1), lq23
Soft tissue sarcoma, osteosarcoma, breast cancer, brain tumor, leukemia, ACC
ACC, 3%
Beckwith-Wiede mann
IGF2, H19, CDKN1C, KCNQ1 (11p15)
Exomphalos macroglossia, gigantism, ACC, nephroblastoma, hepatoblastoma, rhabdomyosarcoma
ACC, 5%
Carney complex
PRKAR1A (17q23-q24) 2P16
Cardiac, endocrine, cutaneous and neural myxomatous tumors, and pigmented lesions of the skin and mucosa
PNAD 90–100%
MEN1
MEN1 (11Q13)
Parathyroid, pancreatic islet cell, anterior pituitary and ACTs
ACT, 55%; ACC, rare
Congenital adrenal
CYP21B (6P21.3)
Adrenal hyperplasia, virilization, salt wasting
ACT, 82%; hyperplasia, hyperplasia 100%
Li-Fraumeni Syndrome (LFS)
LFS is an autosomal dominant familial disease characterized by the early onset of tumors and multiple tumors in affected individuals. ACCs have been reported to occur in 3–4% of patients with LFS [4]. Seventy percent of LFS cases are a result of a germline mutation in the TP53 gene [5]. A second variant is caused by a heterozygous germline mutation in the hCHK2 gene [6].
Beckwith-Wiedemann Syndrome (BWS)
BWS is a congenital overgrowth syndrome characterized by exomphalos, macroglossia, and gigantism in the neonate as well as the development of childhood tumors. About 15% of cases with BWS are familial, with the remainder being sporadic. BWS is linked to the 11p15 chromosomal locus. Genes located at 11p15 and implicated in the pathogenesis of BWS are the insulin-like growth factor 2 (IGF2), H19, and cyclin-dependent kinase inhibitor 1C genes [7].
BWS is a congenital overgrowth syndrome characterized by exomphalos, macroglossia, and gigantism in the neonate as well as the development of childhood tumors. About 15% of cases with BWS are familial, with the remainder being sporadic. BWS is linked to the 11p15 chromosomal locus. Genes located at 11p15 and implicated in the pathogenesis of BWS are the insulin-like growth factor 2 (IGF2), H19, and cyclin-dependent kinase inhibitor 1C genes [7].
CNC is a dominantly inherited syndrome characterized by cardiac, endocrine, cutaneous, and neural myxomatous tumors, as well as pigmented lesions of the skin and mucosa [8]. Primary pigmented nodular adrenocortical disease (PPNAD), a main feature of CNC, is a rare cause of adrenocorticotropic hormone (ACTH)-independent Cushing’s syndrome, seen usually in children and young adults.
Multiple Endocrine Neoplasia 1 (MEN1)
MEN1 is an autosomal dominant syndrome characterized by parathyroid, pancreatic islet cell, and anterior pituitary tumors. ACCs have been reported only rarely with MEN1. The MEN1 gene, located on 11q13, encodes the menin protein. Although the function of menin is not known, mutations have resulted in the loss of its function, suggesting that menin has suppressor activities [9].
Congenital Adrenal Hyperplasia (CAH)
CAH is an autosomal recessive disorder resulting from an enzyme deficiency in the cortisol synthesis pathway. Typically, the enzyme 21-hydroxylase, which is encoded by cytochrome P450, family 21, subfamily B (CYP21B), is deficient. A lack of this enzyme leads to compensatory stimulation of the adrenal cortex by corticotrophin-releasing hormone and ACTH with consequent adrenal hyperplasia and overproduction of cortisol precursors, engendering higher levels of androgens. Rarely, ACCs have been described in CAH [10].
Genetics of Sporadic ACC (Table 10.2)
TP53 Gene
The TP53 gene is a tumor suppressor gene that is most frequently mutated in human cancers [11]. The p53 protein is a transcription factor in regulation of the cell cycle, causing cell-cycle arrest or cell death in response to DNA-damaging agents such as radiation and viruses. TP53 mutation is thought to be a late event in the evolution of malignant transformation in sporadic ACTs. Mutations in exons 5–8 of TP53 have been found in 20–27% of sporadic ACCs and 0– 6% of sporadic adrenocortical adenomas (ACAs).
Table 10.2
Evidence of genes involved in sporadic ACTs
Gene (Chromosomal Locus) | Evidence of Involvement in Sporadic ACTs |
|---|---|
TP53 (17p13) | Mutation of TP53 in 20–27% of ACCs and 0–6% of ACAs; 17p13 LOH occurs in ≤87.5% of ACCs and ≤30% of ACAs |
IGF2 (11p15) | Overexpression of IGF2 mRNA in ACCs compared with ACAs. 11p15 LOH occurs in ≤83% of ACCs and 34% of ACAs |
PRKAR1A (17q23-q24) | LOH of 17 q23-q24 occurs in 53% of ACCs and 23% of ACAs; mutation of PRKAR1A occurs in 10% of ACAs but not in ACCs |
MEN1 (11q13) | LOH of 11q13 occurs in 100% of ACCs and 25% of ACAs; MEN1 mutation occurs in 7% of and ACAs |
GNAS (20q13.2) | Mutation of GNAS occurs in ACAs and tumors of patients with adrenocorticotropic hormoneindependent macronodular hyperplasia |
IGF2, p57kip2 (CDKN1C), and H19 Genes
Rearrangements, loss of heterozygosity (LOH; loss of one of two alleles of a gene), and abnormal imprinting of the 11p15.5 locus result in low p57kip2 and H19 and elevated IGF2 mRNA expression, and have been reported in sporadic ACCs. Higher IGF2 expression is associated with a more malignant phenotype, and over-expression of IGF2 is associated with a higher risk of ACC recurrence. Furthermore, LOH of the 11p15 locus has been demonstrated more frequently in ACCs than in ACAs (in 67% of ACCs vs 13% of ACAs) [12].
MEN1 Gene
Because LOH of 11q13 occurs in ≈20% of sporadic ACTs, and adrenal tumors occur in ≤40% of patients from MEN1 kindreds, MEN1 was considered to be a prime candidate gene in the pathogenesis of these lesions. However, on basis of other studies, at present it is unlikely that the MEN1 gene plays a prominent part in the pathogenesis of sporadic ACTs [13].
RKAR1A Gene
PRKAR1A is the main mediator of cyclic adenosine monophosphate (cAMP) signaling. One study found LOH of 17q22–24 (the locus for PRKAR1A) in 23% of ACAs and 53% of ACCs [14].
ACTH-cAMP-PKA Pathway
The binding of ACTH to its receptor (a member of the G protein-coupled receptor family) results in the dissociation of the heterotrimeric Gs, causing the stimulation of adenylate cyclase, which in turn leads to the production of cAMP from ATP. The ACTH-adenylate cyclase signaling pathway has been implicated in the pathogenesis of ACTs in many ways. First, activating mutations of components of the adenylate cyclase pathway have been found in other human endocrine disorders, including toxic thyroid adenomas and acromegaly. Second, there is a correlation between circulating ACTH levels and the size of the adrenal cortex, as seen in patients with CAH or Cushing’s disease [15].
Wnt Pathway
The Wnt family comprises a group of highly conserved growth factors with similar amino-acid sequences, which have roles in developmental and homeostatic processes. The central event in the canonical Wnt signaling pathway is accumulation of beta-catenin in the cytoplasm with subsequent translocation into the nucleus. The Wnt pathway has been implicated in the pathogenesis of several cancers, particularly in patients with familial adenomatous polyposis and in the development of colorectal carcinomas. Because beta-catenin mutations have been found in ACAs and ACCs, it may be an early step in a common multistep pathogenesis of ACAs and ACCs [16].
Molecular Markers in ACC (Table 10.3)
Several studies have assessed the use of immunoistochemical (IHC) molecular markers for discriminating ACCs from ACAs. The markers studied have included insulin-like growth factor (IGF)2, Ki-67/MIB1, p53, murine double minute 2, p21, p27, cyclin D1, Bcl-2, topoisomerase II, human epidermal growth factor receptor 2/neu, E-cadherin, and the retinoblastoma gene product. Many of these molecular markers, however, lack specificity to achieve discrimination between ACCs and ACAs.
Table 10.3
Molecular markers for adrenocortical carcinoma
Marker | Feature |
|---|---|
IGF2/MIBI1 | Yields a sensitivity of 100% and a specificity of 95.5% for differentiating ACCs from ACAs |
Ki-67/MIB1 | Poor sensitivity and specificity |
p53 | Poor sensitivity and specificity |
Murine double minute 2 | Poor sensitivity and specificity |
p21 | Poor sensitivity and specificity |
p27 | Poor sensitivity and specificity |
cyclin D1 | Poor sensitivity and specificity |
Bcl-2 | Poor sensitivity and specificity |
Topoisomerase II | Poor sensitivity and specificity |
Human epidermal growth factor receptor 2/neu | Poor sensitivity and specificity |
E-cadherin | Poor sensitivity and specificity |
Retinoblastoma gene product | Poor sensitivity and specificity |
SF1 (transcription factor) | Can distinguish between primary and secondary ACC |
GATA6 (transcription factor) | ACTs with a Weiss score of 4–9 are lower than the others |
VEGF | Not yet in clinical use |
Combining IGF2 and MIB1 (a mouse monoclonal antibody that recognizes a formalin-fixation resistant epitope on the cell proliferation-associated antigen Ki-67), IHC analyses yielded a sensitivity of 100% and a specificity of 95.5% for differentiating ACCs from ACAs [17].
Transcription factors have also been used as possible molecular markers to differentiate ACCs from ACAs. A member of the nuclear receptor family of transcription factors, steroidogenic factor 1 (SF1) maps to 9q33.3. IHC analyses with SF1 have not been shown to differentiate between ACCs and ACAs, but are useful for distinguishing between primary ACC and metastasis from other sites [18].
GATA6 is from the GATA family of transcription factors, which is characterized by binding to the DNA consensus sequence (A/T)GA T A(A/G). GATA6 has a role in the maturation and differentiation of cells. GATA6 protein expression has been found to be significantly lower in ACCs than in ACAs upon IHC anlyses. Accordingly, ACTs with Weiss scores of 4–9 had a significantly lower GATA6 level than ACTs with Weiss scores of 1–3 [19].
Vascular endothelial growth factor (VEGF) plays a pivotal part in the regulation of normal and tumor angiogenesis. Angiogenesis is critical for tumor growth and metastasis. VEGF has been found to be increased in most cancers and is associated with a worse outcome. Serum VEGF levels have also been assessed in patients with ACCs vs patients with ACAs, and they were not found to be significantly different between the two groups. VEGF, however, as a molecular marker for ACC, has not been integrated into clinical practice [20].
In conclusion, progress into the elucidation of the genes and pathways involved in the pathogenesis of ACC has been slow largely because of the rarity of this tumor. The TP53, IGF2, H19, p57 kip2, and MEN1 genes are involved in adrenocortical carcinogenesis, as are the ACTH-cAMP-PKA and Wnt pathways.
Clinical Presentation (Table 10.4)
ACCs are functional in ≈60% of cases, more commonly in children (85%) than in adults (15–30%). Unlike ACAs that predominantly secrete cortisol, ACCs secrete various hormones, including androgens, cortisol, estrogens, and aldos-terone. In adult patients with functioning tumors, 30% present with Cushing syndrome, 20% with virilization, and 10–20% with a combination of the two. Feminization and hyperaldosteronism are much rarer, each accounting for ≈2% of ACC cases. The rapid onset of Cushing syndrome, often with virilizing features, is characteristic of ACCs in adults. Although benign adrenocortical tumors tend to secrete a single class of steroid, ACCs can secrete various types contemporarily; co-secretion of cortisol with androgens is a frequent combination and is highly suggestive of malignancy. In children, ACCs can present with virilization, Cushing syndrome, feminization, or Conn syndrome.
Table 10.4
Clinical picture of adrenocortical carcinoma
Non-functioning | Clinically silent |
Abdominal mass at palpation | |
Late appearance of compression signs signs due to metastases paraneoplastic signs | |
Functional | Cushing syndrome |
Conn syndrome | |
Virilization (women) | |
Gynecomastia and impotence (men) | |
Feminization and early pseudo-puberty (children) |
The rapid development of hypercortisolism influences its clinical pattern: the main features are profound muscle weakness, skin atrophy, diabetes mellitus and hypertension with hypokalemia (due to activation of mineralocorticoid receptors induced by high levels of cortisol). In patients from the German ACC Registry, autonomous cortisol secretion, either alone or in combination with other steroids, was detectable in 60% of cases in whom hormonal analyses had been unertaken before surgery. However, autonomous cortisol secretion was not clinically suspected in all of these cases. Hypersecretion of adrenal androgens determines virilization in affected women (Fig. 10.1) with amenorrhea, hirsutism, acne and baldness. Estrogen-secreting adrenal tumors in males lead to gynecomastia and testicular atrophy, and are almost invariably malignant. In children, androgen excess determines incomplete iso-sexual precocious puberty in males and incomplete heterosexual precocious puberty in females. In the case of estrogen-secreting ACCs, men suffer from impotence, loss of libido, gynecomastia and testicular atrophy, whereas menstrual disturbances are the main characteristics in fertile women. In children, estrogen excess determines sexual precocity (incomplete hetero- or isosexual precocious puberty in males or females, respectively). Mineralocorticoid-producing ACCs are rare and characterized by hypertension and hypokalemia.
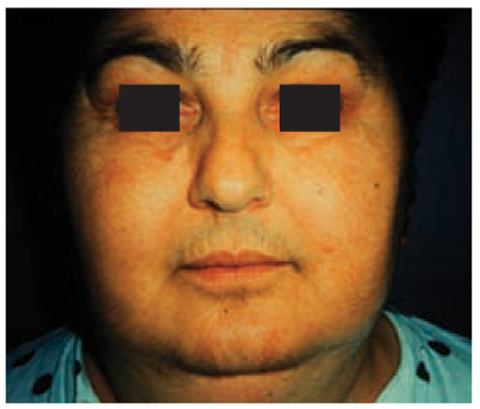
Fig. 10.1
Appearance of virilization in a female patient with an adrenocortical carcinoma
Approximately 65–85% of ACCs in adults are non-functioning, and patients present with a large mass and symptoms related to mass effect (e.g., abdominal or flank pain in 55%) or with a palpable mass (40–50%). Only occasionally do hormonally inactive ACCs present with abdominal discomfort (nausea, vomiting, abdominal fullness) or back pain caused by a mass effect of the large tumor; patients present with fever, weight loss, and anorexia, and it is a remarkable feature of non-cortisol-producing ACCs that wellbeing is often little affected by even a large tumor burden.
Some ACCs are discovered incidentally (0–25%) and tend to be smaller. Due to the late presentation of non-functioning tumors, ≈ 30% of ACC cases present with metastatic disease to the regional and para-aortic lymph nodes, lung, liver, and bone. [21]. In the Italian survey on adrenal incidentaloma, pain was significantly associated with ACCs and was not fully explained by large tumor size per se [22].
A high concentration of dehydroepiandrosterone sulfate (DHEA-S) is another clue suggesting ACC, whereas decreased serum DHEA-S concentrations are suggestive of a benign adenoma. Aldosterone-producing ACCs present with hypertension and pronounced hypokalemia. However, severe hypokalemia is more likely caused by grossly elevated cortisol secretion, leading to insufficient inactivation of renal cortisol by 11-hydroxysteroid dehydrogenase type 2 with consecutive activation of the mineralocorticoid receptor. In many patients with a seemingly hormonally inactive ACC, high concentrations of steroid precursors such as androstenedione or 17-hydroxyprogesterone can often be demonstrated, thereby establishing the adrenocortical origin of the tumor.
Diagnosis and Differential Diagnosis
Hormonal Workup
Careful endocrine assessment is essential before surgery in ACCs (Table 10.5). The pattern of hormone secretion may point to the malignant potential of the lesion (e.g., estradiol in males, high concentration of serum DHEA-S, or secretion of steroid precursors) and may thus affect surgical strategy (open instead of minimally invasive surgery). In addition, autonomous cortisol secretion by the tumor is associated with the risk of postoperative adrenal insufficiency. Due to the variable hypercortisolemia and the rapid development of ACC, the clinical features of Cushing’s syndrome are often incomplete or even absent (atypical or subclinical Cushing’s syndrome). To establish tumor markers for monitoring tumor recurrence, a thorough hormonal workup is essential. Finally, it is important to exclude a pheochromocytoma before surgery because imaging often cannot reliably differentiate between ACC and pheochromocytoma.
Table 10.5
Recommended hormonal evaluation in suspected or proven adrenocortical carcinoma
Cortisol excess (minimum 3 or 4 tests) | |
Dexamethasone suppression test (1 mg, 23.00 h) | |
Urinary free cortisol (24-h urine) | |
Basal cortisol (serum) | |
Basal ACTH (plasma) | |
Sexual steroids and steroid precursors | |
Dehydroepiandrosterone sulfate (DHEAS) (serum) | |
Androstenedione (serum) | |
Testosterone (serum) | |
17-OH-progesterone | |
17-beta-estradiol (serum, in men and postmenopausal women) | |
Mineralocorticoid excess | |
Potasium (serum) | |
Aldosterone: reinin ratio (only in patients with arterial hypertension and/or hypokalemia) | |
Exclusion of pheochromocytoma (minimum 1 or 3 tests) | |
Catecholamine excretion (24-h urine) | |
Metanephrine excretion (24-h urine) | |
Meta- and nor-metanephrine (plasma) |
Imaging
General Properties
Metastatic disease is definitive of malignancy [23]. However, several imaging features should increase the suspicion of ACC within an adrenal mass [24]: tumor diameter >4 cm, irregular tumor margins, central intratumoral necrosis or hemorrhage, heterogeneous enhancement, invasion into adjacent structures, venous extension (renal vein or inferior vena cava (IVC)), and calcification. ACCs are usually large at presentation, ranging from 2 cm to 25 cm (average size ≈9 cm). Approximately 70% of ACCs are larger than 6 cm. They are bilateral in 2–10% of cases and are slightly more common on the left than on the right. Tumors are frequently hemorrhagic and necrotic, and may contain small areas of intracytoplasmic lipid or fatty regions. Using a logistic regression model, Hussain et al. found tumor size >4 cm and heterogeneous enhancement to be the most important discriminators of malignancy [25]. The existence of intracytoplasmic fat in ACCs has been attributed to cortisol and related fatty precursors in hormonally active tumors. On occasion, pockets of fat may be seen within the mass, indicating coexistent myelolipomatous tissue. Invasion into the IVC has been reported in 9–19% of ACC cases at presentation.
CT (Fig. 10.2)
The typical appearance of ACC on unenhanced CT is of a large, inhomogeneous (but well-defined) suprarenal mass that displaces adjacent structures as it grows [26]. Regions of low attenuation correspond to necrosis pathologically. After the intravenous administration of contrast material, there is inhomoge- neous enhancement of the tumor, typically with greater enhancement seen peripherally and relatively little enhancement seen centrally, because of central necrosis [27]. Measurement of the attenuation of adrenal lesions on unenhanced CT is of great value in distinguishing between benign and malignant masses. Cumulative data obtained for the identification of adrenal adenomas indicate that ACCs rarely have an attenuation <10 Hounsfield units (HU). The specificity of this threshold for the identification of benign adenomas is ≈98%. Equally, ACCs retain intravenous contrast material and have absolute and relative percentage washout of <60% and <40%, respectively, 15 min after contrast administration or <50% and <40%, respectively, at 10 min [28]. Calcification (micro or coarse) is seen on CT in ≈30% of patients with ACC and is usually centrally located (Fig. 10.3). Calcification is rare in adenomas, but is present in ≈10% of pheochromocytomas [29]. Tumor thrombus extending into the IVC at presentation is not rare and is more frequently seen in right-sided tumors. A tumor thrombus within a vein is usually well encapsulated and can often be withdrawn intact from the vein [30]. The cephalad extent of the tumor thrombus can be identified on contrast-enhanced CT or magnetic resonance imaging (MRI).
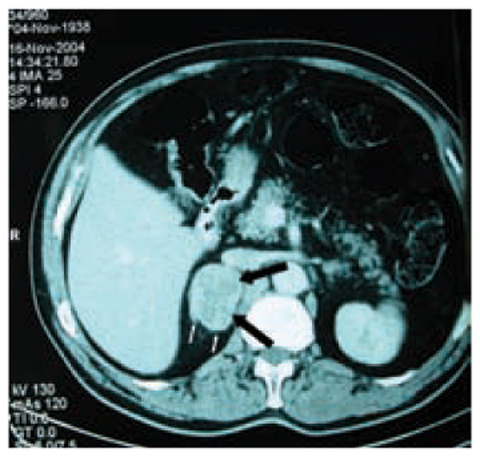
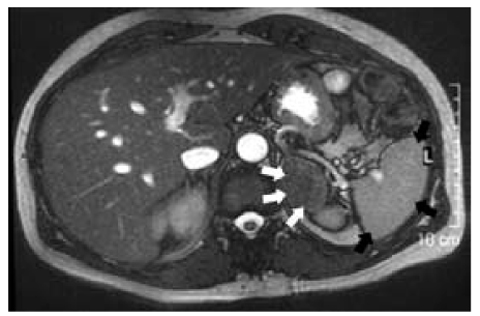
Fig. 10.2
Contrast CT of a right adrenocortical carcinoma. In the arterial phase, an irregularly oblong mass in the right adrenal gland is evident (thick black arrows ). The neoplasm has a main diameter of 5.8 cm, irregular edges, shares some jagged edges (thin white arrows), and a finely irregular structure
Fig. 10.3
MRI features of adrenal malignancy. This left adrenal neoplasm is big, irregularly oblong, and dark in this MRI „out of phase” acquisition. It presents a finely irregular structure, with a „salt and pepper” aspect (arrows) due to necrotic phenomena. All these aspects suggest a malignant adrenal mass
CT is also of value in showing the local and distant spread of an ACC. Preservation of fat planes around the tumor indicates that there is no local invasion. Where there is a paucity of retroperitoneal fat, it may not be possible to determine if the tumor has invaded adjacent organs. Metastases are frequently found at presentation: regional and para-aortic lymph nodes (25–46%), lungs (45–97%), liver (48–96%), and bone (11–33%) are the common sites. Hepatic metastases tend to be hyper-vascular and are best seen on arterial-phase imaging after intravenous administration of contrast.
MRI (Fig. 10.3)
ACCs are typically heterogeneous in signal intensity on MRI because of hemorrhage and/or necrosis [31]. On T1-weighted imaging, ACCs are typically isointense or slightly hypointense to normal liver parenchyma. However, high T1 signal intensity is often seen because of hemorrhage. On T2-weighted imaging, ACCs are usually hyperintense to liver parenchyma and have a heterogeneous texture because of intratumoral cystic regions and hemorrhage [32]. A functioning ACC can contain small regions of intracytoplasmic lipid, resulting in small non-uniform areas of loss of signal on chemical shift imaging (<30% of the lesion) [33]. Although similar small non-uniform loss can occur in lipid-poor adenomas, the significant uniform signal loss seen in lipidrich adenomas does not occur.
The results of early studies suggested that proton MR spectroscopy may be useful in differentiating adrenal adenomas and pheochromocytomas from adrenal metastases and ACCs. Faria et al. looked at the spectral traces obtained from 60 patients with adrenal masses. Adenomas and pheochromocytomas could be differentiated from ACCs and metastases using choline:creatine ratios of >1.20 (92% sensitivity and 96% specificity) and choline:lipid ratios of >0.38 (92% sensitivity and 90% specificity). ACCs and pheochromocytomas could be differentiated from adenomas and metastases using a 4.0–4.3 ppm/creatine ratio >1.50 (87% sensitivity and 98% specificity). By combining these two spectral analyses, they could divide adrenal mass lesions into one of four distinct groups: adenoma, pheochromocytoma, ACC, or metastasis [34]. Although there were some criticisms of this study, the method appears to offer potential in helping to distinguish among adrenal mass lesions.
Functional Imaging (Fig. 10.4)
Fluorodeoxyglucose-positron emission tomography (FDG-PET) can be used to identify certain malignant adrenal masses by virtue of their increased metabolic activity; however, if FDG uptake is only modest, the likelihood of benign vs malignant is approximately equal [35]. FDG-PET combined with contrastenhanced CT has a sensitivity of 100% and specificity of 87–97% for identifying malignant adrenal masses. The lower specificity is because a small number of adenomas and other benign lesions mimic malignancy [36]. The novel PET tracer 11C metomidate (a marker of 11β-hydroxylase) is used as a tracer for adrenocortical tissue and is taken up by adenomas and ACCs. This marker differentiates adrenal cortical lesions from pheochromocytomas and metastases, which are uptake-negative [37]. However, the most valuable aspect of PET is its ability to detect distant metastases (one-third of patients with ACCs will have metastatic disease at presentation).
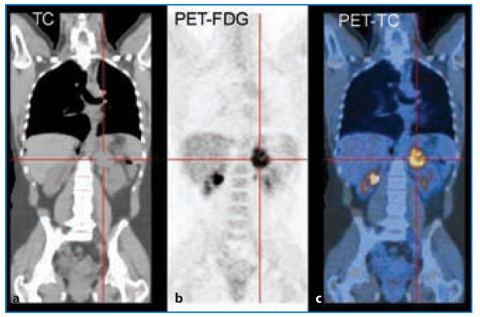
Fig. 10.4




FDG-PET (b) and CT-PET (c) of adrenocortical carcinoma. PET of a small left adrenal incidentaloma is shown. It was iso-dense at CT (a), which is a suspicious criterion of malignancy, so FDG-PET and PET-CT were necessary, even if the mass did not share a dimensional aspect typical of adrenocortical carcinoma (dimension, ≈3.9 cm). The final diagnosis was a small adrenocortical carcinoma at a very early stage (T1 according to the WHO classification)
Related posts:


Stay updated, free articles. Join our Telegram channel
Full access? Get Clinical Tree
