swollen, and quite uneven around the edges. At midday he felt cold, then he became hot, feverish. In the evening, he vomited bitter, biliary fluids. His eyes were teary, light did not bother him very much. The cuts had small blisters in the middle and around the uneven edges. They were fetid. Moderate fever, no thirst. He was kept under watch overnight. Moderate urination with dark yellow sediment.
 FIG. 43.1. Front page of the publication by the Academy of Science of Siena reporting the history of the variolation campaign made in Siena between 1758 and 1760. Reproduction by courtesy of the Accademia de’ Fisiocritici, Siena, Italy. |
vesicle fluid from infected cows. In 1940, Collier developed a modern commercial process to produce a stable freeze-dried vaccine for large-scale smallpox prevention. Because Variola virus is transmitted from human to human and does not have any animal reservoir, it was hypothesized that eradication of smallpox through vaccination was possible. In 1959, the World Health Organization (WHO) set global smallpox eradication as a goal, but it was only in 1966 that a Smallpox Eradication Unit was formed. The global eradication program started in 1967. It is estimated that at that time, there were still 10 to 15 million cases of smallpox worldwide. The vaccination campaign lasted about 10 years and was extremely successful. The last case of naturally contracted smallpox was recorded in 1977 in Somalia, and in 1979, the Global Commission for Smallpox Eradication certified that global smallpox eradication had been achieved.
TABLE 43.1 Development of Human Vaccines | |||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| |||||||||||||||||||||||||||||||||||||||||||||||||
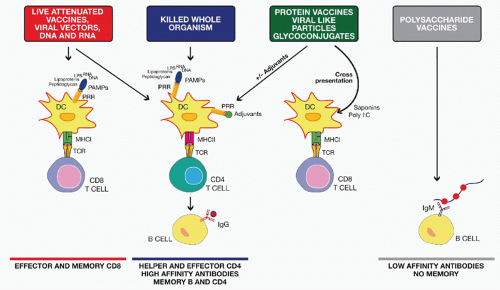 FIG. 43.2. Mode of Action of Different Classes of Vaccines. Live attenuated, viral vectors and nucleic acid vaccines induce both major histocompatibility complex (MHC) class I and MHC class II antigen presentation and activate pattern recognition receptors (PRRs) expressed by dendritic cells (DCs) through several pathogen-associated molecular patterns (PAMPs) including peptidoglycan, lipopolysaccharide, and bacterial or viral ribonucleic acid and deoxyribonucleic acid. They generally induce robust cluster of differentiation (CD)8 and CD4 T-cell responses, high antibody titers, and good memory. Killed whole-organism vaccines activate DCs through the same PAMPs and induce good CD4 and B-cell responses. Protein vaccines, viral-like particles, and glycoconjugate vaccines may need an adjuvant for optimal DC activation and CD4 T-cell priming. Some adjuvants such as polyinosinic:polycytidylic acid or saponins can also induce CD8 T-cell responses to subunit vaccines through cross-presentation mechanisms. Finally, polysaccharide vaccines induce a T-cell-independent antibody response. |
profile. A drawback of a clean vaccine preparation containing pure recombinant protein(s) is their reduced immunogenicity that may require the addition of an adjuvant to achieve enhanced efficacy.
more efficacious, being able to induce a long-lasting antibody response not only in adults but also in infants, young children, and the elderly. Similarly to the polysaccharidebased vaccines, the glycoconjugate vaccines are also serotype specific, and, therefore, multivalent formulations are required to achieve protection against multiple serotypes.
followed by a boost with a protein-based vaccine, although other options have also been used. This unconventional immunization schedule usually results in the induction of a strong cellular immune response, with generation of both CD4+ and CD8+ antigen-specific T cells. This is in turn associated with a higher and more specific antibody response against the vaccine target compared to homologous immunization with either DNA or viral vector. The immunologic mechanisms responsible for these improved responses are not entirely known. It is hypothesized that while DNA or viral vector priming is required to induce a strong T-cell response, boosting with only the target protein focuses the B-cell response on a single protein antigen for which high-affinity antibodies are desirable and at the same time avoiding interference by antivector immunity.34
T-cell responses and in particular CTLs, in cases of viral infections57 or cancer,58 where these type of immune responses are required to obtain a complete protection. Although the peptide approach is able to induce protection from viral infections and cancer in the mouse model, polymorphism of the MHC system has limited the application of peptides to human preventive vaccines. The use of peptides has been mostly restricted to therapeutic vaccines against cancer.59
Increase persistency of antigen at injection site
Enhanced antigen uptake by APCs
Production of cytokines and danger signals at injection site
Activation of APCs and other cells of the innate immune system
Increased cross-presentation by DCs
Recruitment of DCs and other immune cells to the injection site
Migration of antigen-positive APCs to draining lymph nodes
More efficient B-cell receptor engagement by making antigen multivalent
Activation of B-cell proliferation and differentiation into plasma cells
adjuvanted influenza vaccines; and AS04, a combination adjuvant composed of the TLR4 agonist monophosphoryl lipid A (MPL) adsorbed to alum, has been approved for HBV and HPV vaccines. The identification and development of new adjuvants is necessary because the limited number of currently approved vaccine adjuvants do not always elicit the desired immune responses capable of preventing the targeted infection. In particular, there is a need for new adjuvants eliciting potent CD8+ T-cell and Th1 responses. A large number of compounds have demonstrated vaccine adjuvant activities in preclinical models, and it would be difficult to review all of them in this chapter. Therefore, we will focus only on the adjuvants that have been either licensed (Table 43.2) or already tested in human (see Table 43.3), with a special focus on their use with subunit vaccines targeting infectious diseases.
TABLE 43.2 Licensed Human Vaccine Adjuvants | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
oil, emulsified with two surfactants (Tween 80 and Span 85). MF59 has been licensed in Europe since 1997 in an adjuvanted seasonal influenza vaccine for its capacity to increase flu immunogenicity in the elderly.84 More recently, MF59 and a second squalene-based oil-in-water emulsion called AS03, composed of squalene, Tween 80 and α-tocopherol, have been licensed in Europe for pandemic influenza vaccines and have been widely used for the 2009 H1N1 pandemic flu campaign.85 A third squalene-based oil-in-water emulsion called AF03 has been approved for pandemic influenza in Europe but has not been marketed yet.86 Clinical trials demonstrated that avian H5 pandemic influenza vaccines containing oil-in-water emulsions are superior to alum-adjuvanted or nonadjuvanted H5 vaccines in the induction of protective antibody titers.87 Both AS03 and MF59 allowed for antigen dose sparing and increased seroconversion and cross-protection.88,89 It is important to notice that in the case of H5 avian flu, for which most of the population has not been previously exposed, nonadjuvanted vaccines did not reach the antibody titers considered to be the serologic correlate of protection, whereas emulsion-adjuvanted vaccines were able to elicit protective antibody titers after two doses. A phage display library approach demonstrated that MF59 can enhance the breadth and the affinity of the antibody responses to H1 and H5 pandemic influenza vaccines.90,91,92 In particular, the addition of MF59 enhanced the recognition of the HA globular head region that contains most of the neutralizing epitopes. Two additional clinical trials have been conducted on individuals vaccinated several years before with H5 vaccines either nonadjuvanted or mixed with MF59 or AS03 by administering a new dose of oil-in-water emulsion-adjuvanted H5 vaccine (boost). These studies demonstrated that priming with avian H5 vaccines mixed with MF59 or AS03 allows for better and more rapid responses to a booster dose compared to individuals primed with nonadjuvanted vaccines.93,94 Priming with adjuvanted vaccines was effective even when H5 antigens from different influenza clades were used for priming and boosting, inducing a rapid increase of cross-neutralizing antibody titers. These studies suggest that adjuvanted influenza vaccines may be used for a prepandemic vaccination strategy. Another recent study highlighting the strong impact that adjuvants can have on vaccine efficacy was recently performed on children between 6 and 72 months of age. The addition of MF59 to seasonal flu subunit vaccine increased the efficacy of the vaccine from 43% to 86% against polymerase chain reaction confirmed influenza-like illness.95 Interestingly, in the age subgroup 6 to 24 months, nonadjuvanted influenza vaccine was unable to protect, whereas the efficacy of MF59-adjuvanted vaccine was 77%.
 -tocopherol in the formulation.
-tocopherol in the formulation.signals for naive T-cell priming and development of an adaptive immune response. In addition to pathogen infection, microbial products from live-attenuated or heat-killed vaccines can also stimulate TLR pathways providing adjuvant activities.105,106 For example, the yellow fever vaccine activates DCs by signaling through TLR2, 7, 8, and 9.107 Many preclinical and clinical studies using purified TLR agonists have been conducted and demonstrated that all TLRs examined can be exploited to enhance adaptive responses to vaccination. However, despite the large amount of work performed, so far, only the TLR4 agonist 3-O-desacyl-4′-MPL, a component of lipopolysaccharide (LPS) purified from Salmonella minnesota has been licensed for human use. MPL is approved for two vaccines against HPV and HBV. In both vaccines, MPL has been adsorbed to alum to form a combination adjuvant called AS04. The HBV vaccine formulated with AS04 was developed for patients with renal insufficiency and was tested in several immunocompromised individuals including the elderly and patients on hemodialysis. The incorporation of MPL allowed for a more rapid increase of antibody titers and enhanced seroprotection rates using fewer vaccine doses.108 HPV vaccine formulated in AS04 induced higher neutralizing antibody titers and memory B-cell responses compared to the same vaccine formulated only with alum.109 A head-to-head trial was conducted to compare the alum-MPL adjuvanted HPV vaccine versus another HPV licensed vaccine that is adjuvanted only by alum.110,111 Both vaccines exploit VLP technologies for the production of antigens and are highly efficacious in preventing HPV16/18-associated cervical intraepithelial neoplasia in women. However, the AS04-adjuvanted vaccine produced significantly higher neutralizing antibody titers to HPV16 and HPV18, and higher frequencies of memory B cells compared to the other vaccine.
TABLE 43.3 Vaccine Adjuvants in Clinical Development | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Mycobacterium tuberculosis (Mtb) antigens Ag85B-ESAT-6 and tested in a phase I clinical trial.129 IC31 significantly boosted antigen-specific IFN-γ-producing T cells.
cost-effectiveness. Out of five licensed mucosal vaccines, four are live attenuated (intranasal flu, oral rotavirus, OPV, and typhoid) and one is made of killed V. cholerae given with cholera toxin (CT) B antigen. Notably, no subunit vaccine has been licensed for mucosal administration due mainly to the absence of safe and effective human mucosal adjuvants. In fact, the administration of antigens in the absence of adjuvants in respiratory or gastrointestinal tracts can induce tolerance. Mucosal adjuvants are required to 1) protect antigens from degradation by mucosal proteases, 2) enhance internalization of vaccine antigen by intraepithelial DCs or M cells, and 3) overcome regulatory mechanisms that prevent the development of effector B- and T-cell responses in the mucosal-associated lymphoid tissue. Mucosal immunity can protect from pathogen infection by various mechanisms including local production of IgA, IgG, and IgD antibodies by mucosal B cells; activation of mucosal CD4+ T cells producing IL-17 (Th17); and activation of mucosal CD8+ T cells.146 A large number of animal studies showed that some TLR agonists such as CpG ODNs, MPL, and flagellin can act as mucosal adjuvants; however, they have not been tested in humans yet.147 The most advanced mucosal adjuvants are CT and heat-labile toxin (LT) from E. coli
Related posts:






Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


