Neuroblastoma
Neuroblastoma is an enigmatic malignant neoplasm. In its early stages it can be readily cured with surgery or, in some circumstances, can even spontaneously regress or mature to a benign ganglioneuroma. In the more common advanced stages, the disease is often fatal. The unique biology of neuroblastoma has attracted the interest of many prominent scientists and is one of the first malignancies in which molecular biologic assays have influenced treatment and prognosis.
 EPIDEMIOLOGY
EPIDEMIOLOGY
After brain tumors and leukemia, neuroblastoma is the third-most-common malignancy diagnosed in children, with an annual incidence of 9 new cases per 1 million children in the United States. It is the most common cancer diagnosed before the age of 12 months, accounting for almost half of all cancers in infants. The median age at diagnosis is 2 years. The relatively good prognosis of infants diagnosed with early-stage neuroblastoma prompted the initiation of infant-screening studies.1
Screening of infants for neuroblastoma has been studied systematically in large clinical trials conducted in Japan, North America, and Europe.2–5 Excretion of catecholamine metabolites in the urine of children with neuroblastoma has served as the basis of these screening tests. Urine samples were collected and dried on filter paper and returned to screening centers to be tested qualitatively for vanillylmandelic acid (VMA) levels or quantitatively for VMA and homovanillic acid (HVA) measured by high-performance liquid chromatography and normalized to urinary creatinine levels. Children with elevated levels of these metabolites subsequently were referred for further diagnostic evaluation. The incidences of the diagnosis and death rates from neuroblastoma in the screened populations were compared to control populations in the same country or continent. The study populations generally were chosen because of access to an existing screening infrastructure that could readily be adapted to neuroblastoma. The Japanese study screened children at 6 months of age. The Quebec study screened children at 3 weeks and 6 months of age. The German study tested children at their first year’s birthday.
The results and conclusions of these three large screening trials on three continents are remarkably similar. In Japan, 1,142,519 children were screened using a qualitative test, and another 550,331 were screened with the quantitative test; they all were compared to 713,025 children in a control population. The incidence rates per 100,000 were 1.12 in the control group compared to 5.69 in the qualitative group and 17.81 in the quantitative group. Despite this increased incidence in the screened group, the neuroblastoma mortality rates were unchanged by the screening.5 In Germany, 1,475,773 children were screened between 1994 and 1999. Screening detected neuroblastoma in 149 children, 3 of whom died. Despite the screening at 12 months, another 55 children subsequently developed neuroblastoma, 14 of whom died. Compared to the control group, the incidence of stage 4 neuroblastoma was similar. The death rate from neuroblastoma was unaffected by the screening.3 In Quebec, a total of 425,838 children were enrolled in the neuroblastoma screening trial (89% of births during a 5-year study period from 1989 to 1994). The standardized incidence ratios of neuroblastoma death in the Quebec cohort were nearly identical to those in control groups in Ontario, Minnesota, Florida, and the Greater Delaware Valley.4 In summary, each of these trials demonstrates that screening does not affect the mortality rate of neuroblastoma. Furthermore, the overdiagnosis of clinically insignificant disease may lead to significant financial, emotional, and physical burdens on the children and their families. This tumor has a high frequency of spontaneous regression in infancy. The mass screening programs have led to the diagnosis of biologically favorable and clinically insignificant tumors.6,7
 NATURAL HISTORY
NATURAL HISTORY
Neuroblastoma, along with ganglioneuroma and ganglioneuroblastoma, may arise from any site in the sympathetic nervous system. The most common sites of origin are the adrenal medulla (30% to 40%) and paraspinal ganglia in the abdomen or pelvis (25%). Thoracic (15%) and head and neck primary tumors (5%) are slightly more common in infants than in older children. More than 70% of patients have metastatic disease at presentation. The most frequent metastatic sites are lymph nodes, bone, bone marrow, skin (or subcutaneous tissues), and liver.8 The lung and central nervous system are rare sites of involvement.
Neuroblastoma has the highest spontaneous remission rate of any human neoplasm, usually by maturation to ganglioneuroma.9 Microscopic neuroblastomas have been found in the autopsy material of the adrenal glands of young infants at >40 times the expected rate. It has been suggested that most potential neuroblastomas are never clinically manifested, because of spontaneous regression.10 Despite these peculiarities, clinically obvious neuroblastoma is frequently a progressive and relentless disease.
 CLINICAL PRESENTATION
CLINICAL PRESENTATION
Pain is the most common presenting symptom. This frequently is caused by bone, liver, or bone marrow metastases or local visceral invasion by the primary tumor. Other constitutional symptoms may include weight loss, anorexia, malaise, and fever. Respiratory distress may accompany massive hepatomegaly, especially in infants with stage IV-S disease.8 Horner’s syndrome can accompany a primary tumor originating in the neck. Spinal cord compression with paralysis of the lower extremities can accompany the so-called dumbbell-shaped tumor that extends from its origin along the sympathetic ganglia through the adjacent neural foramina. Orbital metastases are not uncommon and can cause proptosis and ecchymosis. Skin metastases may have a bluish tinge, giving the classic “blueberry muffin” sign. When pressed, the release of catecholamine into the tissue causes transient blanching of the adjacent skin. An unusual presentation of localized neuroblastoma is the opsoclonus–myoclonus syndrome, manifested by truncal ataxia and cerebellar encephalopathy. This syndrome typically indicates a favorable prognosis from the tumor, but patients may have persistent neurologic sequelae after successful tumor therapy.11,12
 DIAGNOSTIC WORKUP
DIAGNOSTIC WORKUP
As in the case of any suspected malignant neoplasm in children, the diagnosis of neuroblastoma must be established by pathologic evaluation. Tumor tissue may be obtained from the suspected primary tumor site or from involved lymph nodes by excision (if the tumor is resectable) or incisional biopsy. Bone marrow aspirate and biopsy frequently show metastatic tumor deposits that can establish the diagnosis. Pathologic evaluation of bone marrow is also a requirement for staging of neuroblastoma. Characteristically, neuroblastoma in bone marrow appears in clumps and pseudorosettes. The absence of pseudorosettes does not eliminate the possibility of neuroblastoma.
Laboratory studies should include measurement of urinary catecholamines and their metabolites. Either HVA or VMA, metabolites of dopa/norepinephrine and epinephrine, respectively, is elevated in >90% of patients with stage IV neuroblastoma. A ratio of VMA to HVA of >1.5 is associated with a favorable prognosis in patients with metastatic neuroblastoma. An assay for urinary VMA is the basis for screening studies of infants in Japan, Europe, and North America. Anemia secondary to bone marrow involvement with tumor can be evaluated with a complete blood cell count. Serum ferritin, lactate dehydrogenase (LDH), and other liver function indicators should be assayed routinely.
Appropriate use of imaging studies assists in staging and in planning an approach to therapy. X-ray studies demonstrate intrinsic speckled calcifications in 85% of neuroblastomas. Computed tomography (CT) of the abdomen with intravenous contrast is more sensitive than intravenous pyelography and provides more information about lymph-node or hepatic metastases as well as tumor resectability.13 Increasingly, high-quality magnetic resonance imaging (MRI) scans are replacing the routine use of CT in evaluation of suspicious thoracic or abdominal masses in children. Although MRI cannot demonstrate intratumoral calcifications, it allows better evaluation of blood vessel encasement, intraspinal extension (dumbbell tumors), diffuse hepatic replacement, and bone marrow involvement (Fig. 86.1). Each of these findings improves staging accuracy and facilitates the decision-making process regarding appropriate surgical interventions.14–17
Nuclear medicine scans are helpful in determining the extent of metastatic disease. Because neuroblastoma has a predilection for bony metastases, a radionuclide bone scan is a mandatory investigation. It is more sensitive than a skeletal survey in detecting bone metastases18 Meta-iodobenzylguanidine (MIBG) is concentrated by neurosecretory granules of both normal and neoplastic tissues of neural crest origin and can be used to image primary and metastatic sites of neuroblastoma. MIBG labeled with either 131I or 123I has a sensitivity of 85% to 90% and a specificity of almost 95% in the detection of metastatic neuroblastoma.19 Poor scintigraphic response on 123I-MIBG scans after induction chemotherapy has been shown to predict for a poor event-free survival in patients undergoing high-dose chemotherapy with stem cell rescue.20 Like other neural crest–derived neoplasms, neuroblastoma can express somatostatin receptors. The long-acting somatostatin analog octreotide labeled with 123I has been used to image neuroblastoma with a sensitivity comparable to that of 131I-MIBG.21 The expression of somatostatin receptors by neuroblastoma tissues is a favorable prognostic factor.22
The Radiology Diagnostic Oncology Group enrolled 96 children with newly diagnosed neuroblastoma in a multicenter prospective cohort study prior to surgery. CT, MRI, and bone scintigraphy were used to evaluate tumor stage. The results show that MRI is more accurate than CT for detection of stage IV disease (sensitivities 0.83 and 0.43, respectively). When combined with bone scintigraphy, both imaging tests have high accuracy for the detection of metastases. Figures 86.2 to 86.4 illustrate the value of CT and MRI in staging patients with metastatic disease. The prevalence of determinants of local disease was relatively low in this study because patients who had extensive disease at time of entry underwent delayed surgery after induction chemotherapy. Although the numbers are small, the data suggest the following: (a) in stage I tumor, abdominal extent was more likely to be staged correctly with CT than with MRI, which often overstaged tumor; (b) for stage II and III tumors, both CT and MRI were more likely to understage than overstage tumor; and (c) for stage II tumor, understaging was more likely with CT than MRI.
FIGURE 86.1. Magnetic resonance imaging scans of a 4-month-old child with a thoracic neuroblastoma. The dumbbell shape of the paraspinal mass can be appreciated on axial (A), coronal (B), and sagittal (C) images. The intraspinal component is indicated by arrows. The three views of this child’s tumor are helpful in radiation-therapy field design.
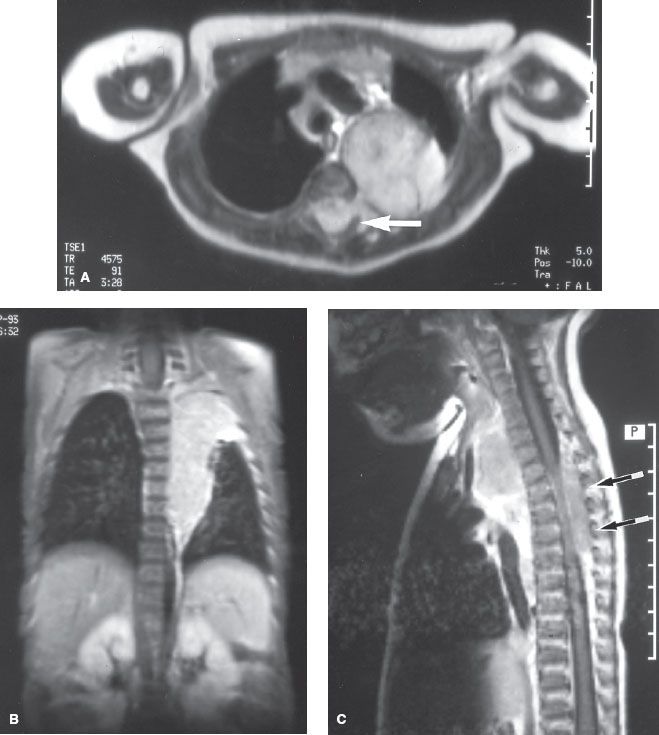
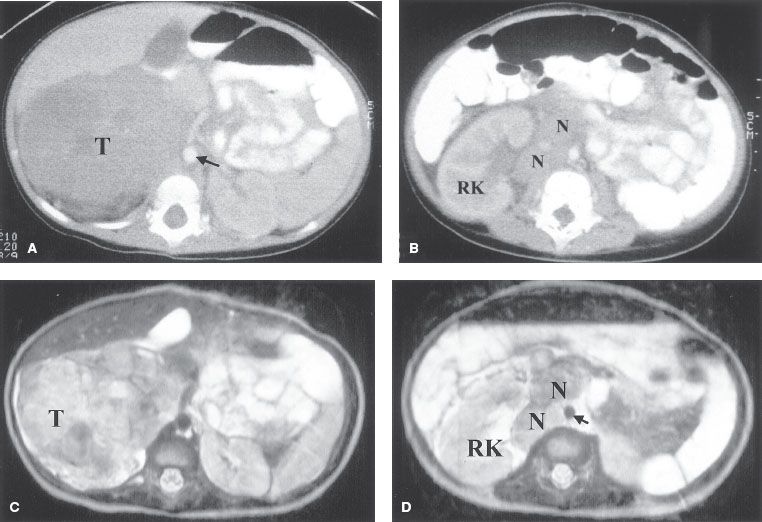
FIGURE 86.2. Neuroblastoma with nodal involvement in a 3-year-old boy. A: Computed tomography (CT) through the upper abdomen shows a large soft-tissue mass (T) arising in the right adrenal gland and extending to the midline (aorta, arrow). B: A CT scan several centimeters lower shows several small nodes (N) in the right pararenal area. C: T2-weighted axial image shows a high–signal intensity tumor (T) in the right suprarenal area. D: T2-weighted image at a lower level shows enlarged, mildly enhancing paracaval lymph nodes (N). (R, right kidney; arrow, aorta). (Courtesy of Dr. Marilyn Siegel, Mallinckrodt Institute of Radiology, St. Louis, MO.)
 STAGING
STAGING
The most commonly used staging system is the International Neuroblastoma Staging System (INSS). It is based on clinical, radiographic, and surgical findings.23 The INSS integrates many of the concepts of previous staging systems promoted by the Children’s Cancer Group (CCG) and Pediatric Oncology Group (POG)24,25 and unifies them into a single system. Each of these systems is summarized in Table 86.1.
 PATHOLOGIC CLASSIFICATION
PATHOLOGIC CLASSIFICATION
Neuroblastomas are derived from primitive neural crest cells arising from within sympathetic ganglia. Three types of tumors, representing different degrees of differentiation, are recognized. Ganglioneuroma consists of mature ganglion cells, Schwann cells, and nerve bundles and is benign in appearance and nature. It frequently is calcified and may represent a matured neuroblastoma.26 Cases of maturation of proven neuroblastomas to ganglioneuromas, either spontaneously or after therapy, have been reported.9 Ganglioneuroblastoma is the intermediate form between ganglioneuroma and neuroblastoma. Both mature ganglion cells and undifferentiated neuroblasts are evident.
Neuroblastoma is at the undifferentiated end of the spectrum of neural crest tumors. It is a small, round, blue cell tumor composed of dense nests of hyperchromatic cells. Homer Wright rosettes with a central fibrillary core can be present. Areas of necrosis, hemorrhage, and calcium are frequently present. Immunohistochemical stains may help distinguish neuroblastoma from other undifferentiated malignant neoplasms of childhood. Neuroblastoma characteristically stains positive for neurofilaments, neuron-specific enolase (NSE), synaptophysin, and chromogranin A and negative for muscle and leukocyte common antigens. The use of electron microscopy to demonstrate neurosecretory granules is required infrequently to establish the diagnosis.
A grading system has been proposed by Shimada et al.,27 and its significance has been confirmed by the CCG.28,29 This clinicopathologic staging system evaluates tumor specimens for stromal development (i.e., stromal-rich and stromal-poor tumors), neuroblastic differentiation, and mitosis-karyorrhexis index of neuroblastic cells. These three histologic features and the patient’s age at diagnosis divide children into favorable and unfavorable prognostic groups. The stroma-rich tumors are characterized by an extensive Schwann cell stroma. The well-differentiated stroma-rich tumors may correspond to ganglioneuroma, and the “intermixed” stroma-rich tumors may correspond to the ganglioneuroblastomas. To be reliable, the Shimada classification requires pretreatment evaluation of the entire primary tumor specimen. However, primary tumors often are not completely resectable, or the presence of widespread metastases makes thorough tumor resection inappropriate before introduction of initial systemic therapy, thereby limiting the usefulness of this system.
FIGURE 86.3. A 1-year-old child with constipation and a palpable mass, also noted to have lower leg weakness. A: Computed tomography shows a large soft-tissue mass with calcifications and necrosis filling the retroperitoneum. Tumor calcification (arrow) is noted in the spinal cord. B: Sagittal, short-tau inversion recovery image shows the large prevertebral mass displacing bowel loops superiorly. Intraspinal tumor extends from the lower thoracic level through the lumbar level. The patient underwent emergent resection of the intraspinal tumor (arrows). (Courtesy of Dr. Marilyn Siegel, Mallinckrodt Institute of Radiology, St. Louis, MO.)
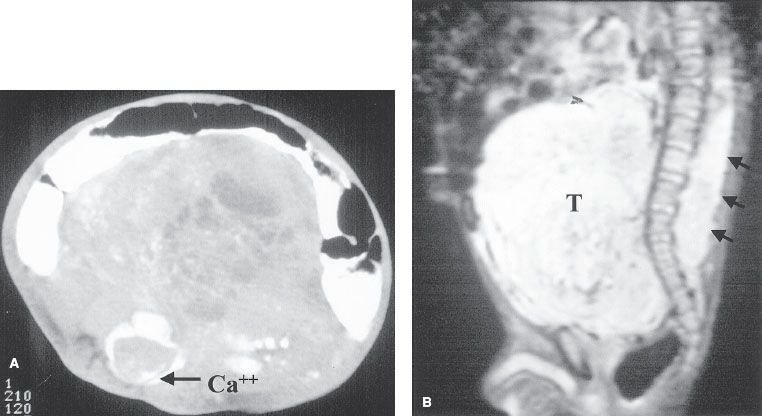
FIGURE 86.4. A 6-year-old boy who presented with bone pain. Axial (A) and coronal (B) T1-weighted image of the pelvis and femurs show diffusely low–signal intensity marrow from metastases (arrows). C: Fat-saturated T2-weighted image of the pelvis. The involved marrow has increased in signal intensity and is now hyperintense to adjacent fat and soft tissue (arrows). D: Fat-saturated T2-weighted coronal image of the pelvis and femurs. Multiple high-signal foci are noted throughout the long bones (arrows). By comparison, normal marrow has a signal intensity similar to that of muscle on fat-saturated images. (Courtesy of Dr. Marilyn Siegel, Mallinckrodt Institute of Radiology, St. Louis, MO.)
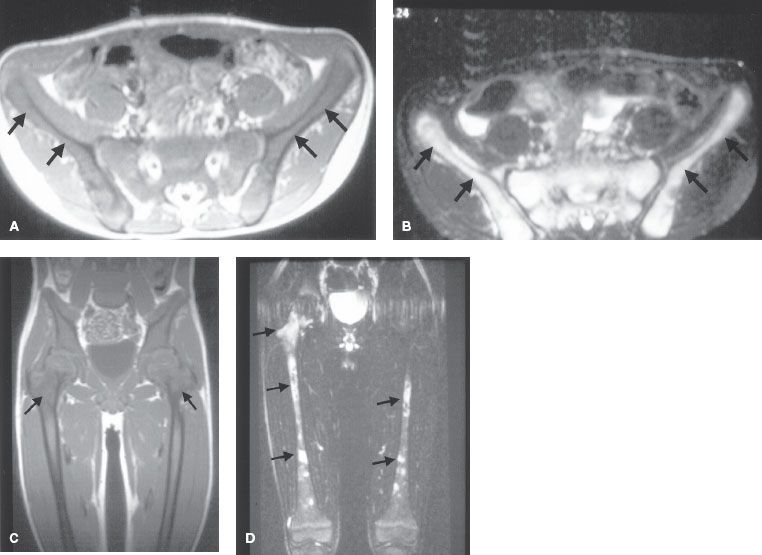
TABLE 86.1 NEUROBLASTOMA STAGING SYSTEMS

 PROGNOSTIC FACTORS
PROGNOSTIC FACTORS
Patient age and stage at initial presentation remain the two most important factors that influence outcome (Table 86.2). In general >75% of infants and children <2 years old survive, as do 90% to 100% of children with INSS stages 1 and 2.30–40 The presence of tumor in regional lymph nodes is a poor prognostic factor and was recognized as such by the POG in their staging system.41 Infants <12 months old with metastatic disease confined to the liver, bone marrow (not bone), or skin (stage IV-S) have a remarkably good prognosis; <75% of these children survive with little or no treatment.8,42 Treatment should be directed at relief of the acute presenting event (often respiratory distress secondary to hepatomegaly), and the temptation to aggressively treat these patients in the absence of other bad prognostic factors should be avoided.
Patients with more-differentiated tumors (e.g., ganglioneuroma, ganglioneuroblastoma) fare better than children with poorly differentiated or undifferentiated neuroblastomas. A favorable Shimada stage is associated with 90% survival, compared with 22% with unfavorable Shimada stages. Elevated serum ferritin (>142 ng/mL), NSE (>100 ng/mL), and LDH (>1,500 IU) are all associated with advanced disease and a poor prognosis.32,43–45
MYCN (N-myc) is a proto-oncogene that resides on the short arm of chromosome 2. An increased number of MYCN gene copies is associated with an extremely poor prognosis (5% survival).46,47 MYCN amplification has been associated with the multidrug-resistance gene and may account for this tumor’s notorious resistance to therapy.48 A tumor with a DNA index of 1 (diploid or near-diploid) paradoxically gives a worse prognosis than tumors that are aneuploid.49 Hyperdiploid tumors occur more often in lower stages and are associated with better chemotherapy responsiveness. Allelic loss of the short arm of chromosome 1 represents a loss of heterozygosity of a tumor suppressor gene and is also associated with a poor prognosis independent of age and stage. It reliably identifies patients with stage I, II, or IV-S disease who have a high risk of relapse and require aggressive therapy.50
TABLE 86.2 PROGNOSTIC VARIABLES IN NEUROBLASTOMA
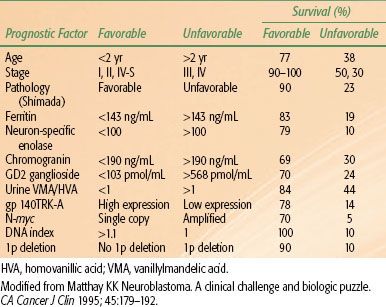
TABLE 86.3 NEUROBLASTOMA RISK ASSESSMENT
 GENERAL MANAGEMENT
GENERAL MANAGEMENT
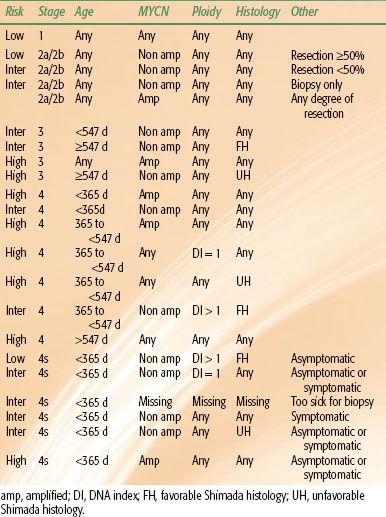
Because of the biologic heterogeneity of neuroblastoma, the following treatment recommendations should be considered as guidelines. The prognostic implications of a tumor’s biologic indices, such as MYCN amplification and DNA index, may warrant more aggressive therapy in young patients with otherwise a favorable stage (Table 86.3).
Low-Risk Disease
Low-stage, resectable tumors (INSS stage 1, 2, or 3 with negative nodes) have an excellent prognosis after complete gross surgical excision. Adjuvant chemotherapy or irradiation has not improved the outcome in children with completely resected tumors with favorable biologic features.38,51–54 Positive surgical margins or microscopic residual disease does not uniformly require more aggressive therapy. Patients with MYCN amplification or low DNA index may require adjuvant therapy and should be enrolled in clinical trials.49
Unresectable tumors that are otherwise of low stage (INSS stages 1 to 3 with negative lymph nodes) may require preoperative chemotherapy and occasionally radiation therapy to convert them into a resectable status. Second-look surgery is performed to remove a previously unresectable primary tumor and achieve a complete remission after induction chemotherapy. Complete resection can be achieved in almost two-thirds of previously unresectable stage III to IV primary tumors.55 The CCG reported that eventual complete resection of the primary tumor in advanced disease may have a favorable impact on outcome.55 The benefit to complete resection in patients with advanced disease has not been uniformly established. Patients with biologically favorable tumors may be more amenable to surgery after chemotherapy, and the apparent benefit of complete resection may be a result of patient selection.56 POG-8104 enrolled patients with INSS stage 1 (POG stage A) disease. In that trial, MYCN amplification and DNA index were not evaluated uniformly. Treatment was surgery only. Regardless of the presence of residual microscopic disease, the 2-year disease-free survival rate was 89%.25 In the CCG experience (CCG trial 3881) with stage 1 disease, the 4-year event-free and overall survival rates for children treated initially with surgery alone were 93% and 99%, respectively. For patients with stage 2 disease, the event-free and overall survival rates were 81% and 98%, respectively. In that trial, only 13% of patients with stage 2 disease received any chemotherapy or radiotherapy, despite the fact that 104 patients had INSS stage 2b disease. The authors ascribe the favorable results to improved surgical management and better staging with MIBG scanning.57
Intermediate-Risk Disease
Locally advanced and regionally metastatic tumors (INSS stage 2b to 3 with positive lymph nodes) require more intensive therapy. Infants <1 year of age should undergo complete resection of the primary tumor and receive adjuvant chemotherapy.56,58–60 In unresectable cases, chemotherapy may be administered initially, and surgery can be performed after response to systemic treatment. In older children with lymph-node metastases, adjuvant radiation therapy to the primary and regional lymph nodes has improved the disease-free and overall survival rates. A prospective, randomized trial of postoperative chemotherapy or chemotherapy plus regional irradiation demonstrated 31% disease-free survival in children treated with chemotherapy, compared with 58% in those who also received radiation therapy.61 However, the value of radiation therapy in intermediate-risk patients is not universally accepted. De Bernardi et al.62 failed to demonstrate a benefit from the addition of radiotherapy in 29 children >1 year of age with postoperative residual tumor or positive regional lymph nodes. Children in that randomized study received two cycles of peptichemio with or without radiation. Progression-free survival was 64% in the radiotherapy arm and 73% in the arm without radiotherapy.62 Coupled with the risk of late effects from even moderate radiotherapy doses, this small trial provided an argument that systematic radiation therapy, even for POG stage C patients, may not be necessary. Current COG trials reflect this bias because the use of radiation therapy is decreasing.
Patients with intraspinal extension of neuroblastoma pose a unique problem. They frequently have severe neurologic compromise resulting from spinal cord compression. Historically, these patients were treated with laminectomy and surgical debulking with or without radiation therapy and chemotherapy.63 Because the morbidity of this approach is significant, with a high rate of spinal growth deformity, a number of investigators have proceeded with treatment of these patients using primary chemotherapy.55 A prospective series of 42 patients treated with primary chemotherapy demonstrated a 92% improvement in neurologic deficits, allowing children to avoid neurosurgical decompression in >60% of cases when receiving courses of carboplatin and etoposide alternating with cyclophosphamide, vincristine, and doxorubicin.
The POG experience (POG trials 8742 and 9244) with stages 2b to 3 disease demonstrates an 85% event-free survival with completely resected tumors at diagnosis, compared to 70% with incomplete resection at diagnosis (p = .259). In both of these studies, patients underwent maximum safe tumor resection followed by five courses of induction chemotherapy. In POG-8742 they received cisplatinum and etoposide alternating with cyclophosphamide and doxorubicin. In POG-9244 they received alternating cycles of vincristine, cisplatinum, etoposide, and cyclophosphamide (OPEC) and vincristine, carboplatin, etoposide, and cyclophosphamide. After second-look surgery the same chemotherapy was given as maintenance. Radiotherapy was given to patients with viable residual tumor discovered at the time of the second-look operation. Children age 12 to 24 months at the time of radiation therapy received 24 Gy in 1.5-Gy fractions to the primary tumor site. Older children received 30 Gy in 1.5-Gy fractions. Of the 37 patients on these two protocols who survived “event free,” 11 (30%) received radiotherapy. Patients with favorable Shimada histology tumors had a 92% event-free survival, compared with 58% with unfavorable tumors (p = .009). Patients with MYCN amplification did poorly, with outcomes comparable to those for stage D patients.27
Based on CCG and POG data, patients with intermediate-risk neuroblastoma have an estimated 3-year survival of between 75% and 98%. Cyclophosphamide, doxorubicin, carboplatin, and etoposide are the four most active agents.64–66 The COG now is evaluating the role of chemotherapy dose intensity and duration for children based on tumor biology.
Surgery plays a critical role in the primary management of neuroblastoma. The goals of surgery are to establish a diagnosis; provide tissue for evaluation of prognostic biologic markers; stage the disease according to INSS criteria; and attempt to totally excise the primary, if feasible. The extent of surgery has an important impact on outcome. O’Neill et al.67 reported that 55 of 59 patients with a complete or near-complete resection were alive and free of disease 2 years after surgery, compared to only 13 of 24 cases with a subtotal resection. Just as Haase had reported, Grosfield and Baehner34 found evidence for improved outcome in stage 4 patients attaining complete resection of primary tumor at delayed second-look procedures.
High-Risk Disease
The majority of patients with neuroblastoma present with metastatic disease. With the exception of infants with favorable biologic disease confined to the skin, bone marrow, or liver, the outcome is poor. Extremely aggressive treatment regimens have been used in these patients to prolong survival and achieve cure. Intensive high-dose chemotherapy regimens appear to be superior to less intensive regimens. Active drugs in advanced neuroblastoma include cyclophosphamide, cisplatin, doxorubicin, etoposide, and teniposide.68
Many recent clinical trials have sought to intensify treatment of metastatic neuroblastoma through the use of high-dose myeloablative chemotherapy with stem cell rescue or bone marrow transplantation (BMT). The French Lyon-Marseille-Curie East group reported a 40% progression-free survival at 2 years and 20% at 5 years in 62 patients proceeding to autologous bone marrow transplant (ABMT).69 The CCG reported a 43% 2-year event-free survival in 43 children undergoing consolidation melphalan, cisplatin, teniposide, doxorubicin, and total-body irradiation (TBI) to 1,000 cGy in three fractions of 330 cGy/day. The toxic death rate was 22%.70 Australian investigators tested a less intense preparative regimen with a TBI regimen consisting of 12 Gy in six twice-daily fractions.71 Of 28 patients registered, 19 achieved complete remission after induction chemotherapy. Seventeen of these 19 patients underwent ABMT and 15 (87%) remained free of disease at 5 years from ABMT. Of the 28 patients registered, 50% have survived 5 years.
Uncertain that ABMT could be studied successfully in a cooperative group, the CCG conducted two pilot studies for children with stage 4 disease. In CCG-321, patients received induction chemotherapy consisting of cisplatin, etoposide, doxorubicin, and cyclophosphamide. Of 207 patients, 159 remained disease free during induction chemotherapy. Of these patients, 67 received myeloablative chemotherapy and ABMT, whereas 74 continued conventional chemotherapy for a total of 13 cycles. The patients receiving the ABMT had a higher event-free survival than patients continuing standard chemotherapy (40% vs. 19%, p = .019).72 Because they are not randomized trials, these studies potentially may have allowed a biased allocation of patients to one arm based on clinical concerns or prognostic risk factors. The POG failed to show a benefit to BMT in high-risk metastatic neuroblastoma (POG 8340).45
The European Neuroblastoma Study Group studied the role of consolidative ABMT with high-dose melphalan versus no further treatment following induction chemotherapy with the OPEC regimen in patients with stage 3 and 4 disease.73,74 With a median follow-up of 14.3 years for surviving children, they report an improvement in event-free survival (38% vs. 27%) and overall survival (47% and 30%) for the high-dose melphalan arm, but these differences did not reach statistical significance. The subset of patients with stage IV disease who were >1 year of age, however, did show statistically significant improvement in event-free survival (33% vs. 17%, p = .01) and overall survival (46% vs. 21%, p = .03).
The CCG conducted a randomized trial comparing continued chemotherapy with myeloablative therapy (including 10 Gy TBI in 3 daily fractions) and autologous bone marrow transplantation in children with high-risk neuroblastoma. All patients received radiation therapy to the primary site and select metastatic sites. A second randomization following cytotoxic therapy included 6 cycles of 13-cis-retinoic acid or no further therapy. Initial results demonstrated an event-free survival advantage to both bone marrow transplantation and 13-cis-retinoic acid therapy, but no overall survival advantage.75 However, after a median follow-up of more than 7 years, a statistically significant advantage to overall survival was demonstrated for the cohort treated with both myeloablative therapy and 13-cis-retinoic acid therapy.76
Berthold et al.77 in Germany reported results of a prospective, randomized trial in children with high-risk neuroblastoma comparing myeloablative therapy with melphalan, etoposide, and carboplatin and autologous stem cell rescue with maintenance chemotherapy with cyclophosphamide. When analyzed as-treated, statistically significantly improved 3-year event-free survival (53% vs. 30%) and overall survival (68% vs. 53%) were seen with myeloablative therapy.
Local control of the primary tumor in stage 4 neuroblastoma is an important element of patient management. The role of surgical resection in metastatic disease remains controversial. Many authors have reported more favorable outcome in patients undergoing complete resection.78,79 There is a strong association between chemotherapy dose intensity and surgical respectability, which reduces the significance of aggressive resection as an independent favorable factor. It can be assumed that tumors that are amenable to resection may have an inherently less aggressive biology.
In the CCG-321-P3 pilot there was a 33% rate of local relapse in patients not undergoing a complete resection at their initial surgery, irrespective of their ultimate surgical resection status, including second-look operations.80 This high local failure rate suggests that there is a role for local radiation therapy. In that pilot study, patients received radiotherapy if they had gross residual disease after second-look surgery. Although the local control in patients receiving radiotherapy was the same as in unirradiated patients, it should be noted that only patients with gross residual disease received this local treatment, suggesting that the radiotherapy (RT) was beneficial.
In a reanalysis of CCG-3891, Hass-Kogan et al.81 compared locoregional control rates in patients on the non-ABMT arm who received 10 to 20 Gy to gross residual disease after induction therapy to patients in the ABMT arm who received similar RT to residual disease but also received 10-Gy TBI in 3.33-Gy daily fractions in their conditioning regimens The patients in the ABMT arm had a lower locoregional recurrence rate (33% vs. 51%, p = .004). Although this affect may be due to the higher dose of RT delivered in this group of patients, it is not possible to separate the RT effect from that of the more intense systemic therapy also given to these patients in the ABMT arm.
Laprie et al.82 analyzed locoregional control in MYCN-amplified INSS stage 2 and 3 patients treated with different regimens over different eras. Their approach varied from conventional chemotherapy and RT only to gross residual disease after surgery in patients >1 year of age, to high-dose chemotherapy with ABMT, to local radiation therapy to all patients. They noted an improvement in event-free survival with ABMT and RT (83% vs. 25%, p = .001). Again, conclusions regarding the effect of RT independent of the intensified systemic therapy are difficult to make because this was not a randomized comparison.
Local therapy to the primary tumor and metastatic sites may be beneficial in the curative therapy of children with disseminated disease. Surgical resection of the primary tumor has been associated with improved survival and local control after aggressive systemic therapy.55,80 There is a predilection for recurrence in previous sites of disease, and it is conceivable that additional local therapy with irradiation to the primary tumor site and distant metastases may enhance tumor control and cure rates.80,83
The POG reported that patients with persistent disease at primary or metastatic sites received boost irradiation of 12 Gy prior to TBI for BMT. Only 2 of 27 patients relapsed at irradiated sites. Six of 10 first-remission patients given local radiation remained in remission, compared to only 13 of 40 who were not irradiated, despite the fact that irradiated patients had residual disease.84
Radiation therapy plays an extremely important role in the palliative management of patients with end-stage symptomatic neuroblastoma. Pain from bone or other visceral metastases often can be relieved with external-beam radiation therapy. Mass effect from a rapidly enlarging tumor can respond dramatically to radiation therapy. In a series of 10 patients treated at Duke University Medical Center, Halperin85 reported 7 complete responses to radiation therapy, either alone or in conjunction with chemotherapy. Radiation doses ranged from 4 to 24.4 Gy at a rate of 1 to 1.5 Gy/fraction. The 7 patients with complete response survived without recurrence.
Systemic radionuclide therapy with 131I-MIBG has been tested in several European and U.S. centers with early encouraging results. This radioactive agent produced objective responses in previously treated and chemoresistant stage 4 neuroblastoma.86,87 These early positive results prompted some investigators to test this agent in previously untreated metastatic neuroblastoma. DeKraker et al.88 reported that a combination of 131I-MIBG and second-look surgery produced response rates comparable to those of multidrug chemotherapy. Preliminary research on the combination of 131I-MIBG with systemic chemotherapy and/or TBI with BMT demonstrated that this is a safe treatment and worthy of more investigation.89–91
The current approach by the Children’s Oncology Group for patients with high-risk disease is to combine induction chemotherapy, surgical resection, autologous stem cell rescue, radiation therapy to the primary site and select metastatic sites, and 13-cis-retinoic acid in an attempt to improve outcome in these patients. The current protocol explores further intensification of therapy by randomizing patients to either one myeloablative transplant or two myeloablative transplants. It will also include a higher radiation total dose of 36 Gy for patients with gross residual disease at their primary sites.
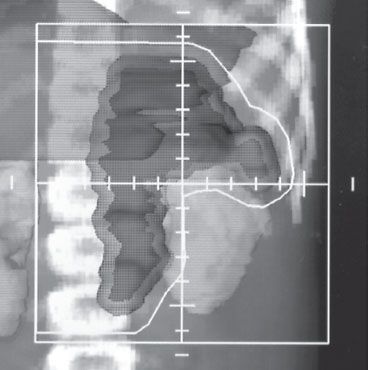
FIGURE 86.5. Radiation therapy treatment field for a child with a left adrenal primary neuroblastoma and para-aortic lymph-node metastases. The beam’s–eye-view display with a digital reconstructed radiograph allows for adequate coverage of the target volume and lymph-node region with sparing of the ipsilateral kidney and liver while homogeneously irradiating the adjacent spine.